Abstract
Jarosite, beudantite and alunite are members of the alunite supergroup. Minerals like those have been detected in different environments on Earth. These jarosite-type compounds are common in acid rock drainage environments and acid sulfate soils, resulting from the weathering of sulfide ores; they are also present in bioleaching systems because they are found in cultures of iron-oxidizing microorganisms. Jarosite is also generated in hydrometallurgical circuits, mainly in zinc hydrometallurgy. These minerals can be used to immobilize different elements such as arsenic and lead, among others. Jarosite and alunite have also been detected on the surface of Mars; the presence of jarosite and alunite and other sulfates provides evidence for the existence of water on Mars. In this work, an exhaustive review of the natural formation, synthesis, structure, thermodynamics, and reactivity of jarosite, beudantite and alunite are included. The capacity of jarosites for the immobilization of the elements, such as lead and arsenic, and information about studies related to jarosite formation on Mars are also included.
1. Introduction
The objective of this paper is to develop a review work about jarosite, alunite and beudantite. This review is focused on the following items: formation of jarosites (natural and synthetic); structure and thermodynamics; reactivity of natural and synthetic jarosites; immobilization of metals in alunite-type structures; passivation of leaching (bioleaching) processes of chalcopyrite induced by the jarosite formation, and the relation between jarosites and Mars.
Jarosites belong to the supergroup of alunites. These minerals are common on the Earth’s surface and consist of three mineral groups with the general formula DG3(TO4)2(OH,H2O)6. D are cations with a coordination number greater or equal to 9, monovalent (Na,K,Ag,H3O+, etc.) or divalent (Pb,Sr,Ba,Ca, etc.). The G site is usually trivalent (Al,Fe). TO4 is dominated by one or more SO42−, AsO43− and PO43−, Dutrizac and Kaiman, 1971 [1], Jambor, 1999 [2], Botinelly, 1976 [3], Scott, 2000 [4], Smith et al., 1998 [5], Viñals et al., 2010 [6] and Sunyer and Viñals, 2011 [7].
The alunite supergroup (with more than 40 mineral species) consists of the alunite group (T position is occupied by sulfates), beudantite group (T position is occupied by one sulfate and one arsenate or one phosphate), and crandallite group (T position one or both are phosphate and arsenate). As seen in the jarosite formula above, the main elements are iron (III) in the G position and sulfur in the T position. Multiple substitutions of different positions are possible, obtaining compounds of good stability. For this reason, some of these compounds have been proposed for the storage of toxic metals [1,2,7]. Examples of compounds corresponding to the supergroup of alunites are included in Table 1. The name “jarosite” includes a set of insoluble compounds of the general formula:
where A can be Ag, ½Pb, Na, K, NH4, Rb, Tl, ½Hg, or hydronium [1]. Only six jarosites have been found as minerals: hydronium (hydronium jarosite), sodium (natrojarosite), potassium (jarosite), silver (argentojarosite), lead (plumbojarosite) and ammonium (ammoniojarosite) [1]. For rubidium (rubidium jarosite), thallium (tallium jarosite), and mercury (mercury jarosite), there is no mineral equivalent and has been chemically synthesized, as shown by Dutrizac and Kaiman, 1976 [1]. These authors detected a metal deficiency in the A position, and an excess of water was observed. This fact indicates the presence of hydronium in the A position. They also indicated a deficiency of iron.
AFe3(SO4)2(OH,H2O)6

Table 1.
Examples of compounds of the alunite supergroup.
The world of jarosites is very wide and its importance appears viewing the high number of publications found in the literature as well as in the review articles carried out by various authors related to jarosite and to alunite or beudantite. Figure 1a shows the number of papers related to jarosite, alunite and beudantite in the last 21 years (Scopus). Figure 1b shows the number of papers related to jarosite, alunite and beudantite in the last 21 years (Web of Science). The number of papers related to jarosite was less than 100 per year between 2001 and 2007; the maximum of papers was 182 in 2020 (Web of Science). The number of papers about alunite was between 29 in 2012 and 79 in 2011 (Web of Science). Finally, the papers about beudantite varied between 0 in 2008 and 12 in 2011.
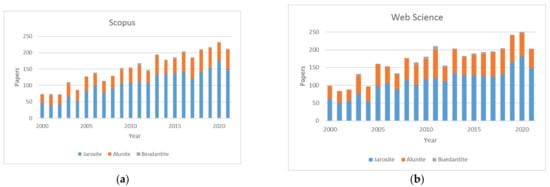
Figure 1.
Number of papers with the terms “jarosite, alunite, beudantite” according to Scopus (a,b) Web of Science.
Jarosite was first described in 1852 by the mineralogist A. Breithaupt in “El Barranco (ravine) del Jaroso”, Almería, Spain, Eftekhari et al., 2021 [8].
Das et al., 1996 [9], carried out a review work about jarosites, focused on the control of Fe (III), sulfates and impurities in hydrometallurgical processes. In this work, aspects related to mineralogy, structure, formation, stability and applications of jarosites are reviewed. The formation of jarosites is favoured between 80–100 °C and pH < 2.5. The presence of mono- and trivalent cations and anions such as sulphate, arsenate, phosphate, silicates, etc., stabilize these compounds even at very high acid concentrations and temperatures > 200 °C.
Eftekhari et al., 2020 [8], developed a review paper in which after indicating that jarosite solids are effective scavengers for metals, inform that these compounds present high precipitation efficiency and ease of filtration. These authors considered aspects of jarosite formation, including their mechanisms, decomposition, biological dissolution, applications and utilization considering the nanoparticles formation, their behavior as catalytic materials, as a fertilizer, pigments, and some other applications.
Hoeber and Steinlechner, 2021 [10], performed an overview of the aspects related to the treatment of iron precipitation residues from the hydrometallurgical zinc industry. Aspects related to iron precipitation, the treatment of these precipitates, considering strategies of immobilization, and processes for the treatment of iron residues, including hydrometallurgy and pyrometallurgy processes are taken into account.
According to Smith et al., 2006 [11], jarosites occur in the oxidised zones of sulfide ore deposits, waters contaminated by an acid rock or acid mine drainage (ARD, AMD), wastes produced from the metallurgical industry, such as in zinc hydrometallurgy, and in acid sulfate soils, among others.
2. Formation of Jarosites
2.1. Natural Jarosites
Natural jarosites have been found in many places on Earth, Hofstra et al., 1999 [12]. Dutrizac and Jambor [13] 2000, indicate that there are four principal modes of occurrence: (1) in the oxidized parts of sulfide deposits or barren pyritiferous rocks, including coal; (2) as nodules and dissemination in clays; (3) as constituents of acid soils; and (4) as hypogene minerals. Some examples of locations where natural jarosites were identified are given here. Argentojarosite was detected at the Tintic Standard mine, Utah (USA); plumbojarosite at Chihuahua (México), and Keno Hill, Yukon (Canada); jarosite and natrojarosite at Shinkolowbwe (Zaire), jarosite in hot-spring deposits at Yellowstone National Park (USA); jarosite at a Kuril Island (Japan); ammonium jarosite at the Sulfur Bank hot-spring mercury deposit, in California (USA), among others.
In the following paragraphs, additional examples of the presence of jarosite, beudantite, and/or alunite in mineral deposits generated by weathering in situ or at some distance from the original sulfide deposits through acid mine drainage are included.
The Iberian Pyrite Belt (IPB) is a huge concentration of massive sulfides located in the SW of the Iberian Peninsula, between Spain and Portugal. In IPB, there are twelve massive sulfide deposits of gossan minerals (Gossan, from English “Gold sand”). Their main components are goethite, hematite, quartz and jarosite. Elements such as sulfur, arsenic, lead, silver and gold, were also detected. Some of these deposits (Tharsis, Rio Tinto, San Miguel, among others) were treated during the last century for recovering gold and silver, Velasco et al., 2013 [14].
Gossan ores from Rio Tinto, Huelva (Spain), were cyanided for about thirty years (1970–2000) for gold and silver extraction by conventional cyanidation. Gold extraction was 75–80% and silver extraction was less than 40%. The chemical and mineralogical composition of gossan was determined to explain the low silver recoveries. The ore consisted of goethite, hematite and solid solutions of beudantite—plumbojarosite—potassium jarosite. Silver was detected as halide, sulfide, mercury/silver sulfo-halide and in jarosite–beudantite phases, Viñals et al., 1995 [15]. These authors indicate that silver extraction by cyanidation corresponded to halide, sulfide and mercury/silver sulfo-halide. The solid solutions of beudantite–plumbojarosite–potassium jarosite were not decomposed during conventional cyanidation in the lime medium. It was the reason for obtaining a silver extraction of less than 40%.
Two types of jarosite were found in gossan ores from Rio Tinto: one corresponding to beudantite, a variable enriched in sulfate; the other is potassium jarosite containing certain amounts of arsenate and lead. Silver is present in both jarosites but is not dissolved by the conventional cyanidation process. The arsenical potassium jarosite can be decomposed in saturated Ca(OH)2 at 70–100 °C. The alkaline decomposition of beudantite was similar but extremely slow at ≤100 °C. The decomposition process includes a slow step of alkaline decomposition followed by a fast step of Ag complexation by cyanide from the decomposition solids, Roca et al., 1999 [16].
Arsenical jarosites were detected in the southern Mother Lode gold district of California (USA). These kinds of compounds were formed by the oxidation of sulfides in mineralized outcrops and mine tailings containing between 100 and 2000 ppm As. The weathering of arsenian pyrite and other As-rich sulfide minerals is conducted to an As retention in the oxidized iron minerals produced, Savage et al., 2005 [17].
Gossan samples from High Lake, Nunavut (Canada), were characterized. Gossan represents a natural acid drainage site in an arctic environment. In two of the outcrops, quartz, mica and jarosite were identified. One of the characterized samples containing Fe-oxide, jarosite and gypsum, appears to be transitional between a Fe-oxide dominant assemblage to a jarosite dominant assemblage. These results indicate that the High Lake gossan deposit does record mechanisms for which minerals like hematite, goethite, gypsum and jarosite, which are found on Mars, can form in an environment that involves seasonal water occurrence in a cold climate, West et al., 2009 [18].
At Mega Livadi, Serifos (Greece), the hypogene mineralization, with pyrite, As-rich tennantite-group minerals and arsenopyrite, underwent strong alteration by descending meteoric waters. Five mineralizing stages were identified. From Stages I to III, oxides, hydroxides and arsenates were obtained: goethite, a mixture of fine-grained Fe–As–Sb–Cu minerals, pharmacosiderite, Sb-bearing beudantite, REE-bearing beudantite, and arseniosiderite, Dill et al., 2010 [19].
Sulphur-bearing minerals such as pyrite are associated with a wide variety of metal ore deposits around the world, examples of which are copper and nickel. Tailings generated in mine processes can produce adequate conditions for jarosite formation, Reynolds 2007 [20]. These residues can contain arsenic and/or silver and gold. In some cases, tailings are subjected to an extractive process for noble metals recovery.
The Veladero high-sulfidation Au–Ag deposit is in the Andean cordillera (Argentina). Volcanic rocks were subjected to acid leaching by weathering, resulting in afine-grained groundmass quartz. Alunite was present there. High-fineness native Au grains were also hosted in quartz. The silicified rocks host Fe oxide/hydroxide and jarosite. According to these authors, jarosite, was the principal host for Ag in Veladero ore, explaining the low (10%) Ag recovery from the oxide ore, Holley et al., 2017 [21].
Tailings from the coal industry can contain jarosites. For example, in the Santa Catarina State coal industry (Brazil), from the run-of-mine, coal-cleaning rejects are obtained. The major minerals were kaolinite, quartz, illite–smectite, pyrite, jarosite and gypsum, among others. Pyrite is relatively abundant in most samples, making up to 5% of the mineral matter. The principal sulfate minerals, jarosite, melanterite, formed due to exposure to the material. Trace metals are also present (Zn, Cu, Pb, As, etc.). Mobilization of these elements as the pyrite (and other sulfides) breaks down during storage may represent a source of adverse environmental impact around such coal cleaning rejects emplacement sites, Cutruneo et al., 2014 [22].
In the Cu–Zn Penn Mine in a massive sulfide belt of the Sierra Nevada, California (USA), acid mine drainage was formed by the oxidation of sulfides exposed on waste piles. The oxidized minerals were secondary Fe minerals, identifying copiapite and jarosite, surrounded by goethite and hematite. This represents the evolution of acid solutions discharged from the pyritic waste piles and the subsequent accumulation of secondary precipitates by hydrolysis reactions, Montero et al., 2005 [23].
The formation of schwertmannite (an iron oxyhydroxide), was investigated in the acid discharge of the Monte Romero mine in the IPB (Spain). This mineral was formed from acid rock drainage (ARD). The mineral precipitated from supersaturated solutions and subsequently transformed into goethite and hydronium jarosite. The formed precipitates from ARD, such as jarosite, schwertmannite and goethite, may play a key role in the removal of trace elements from solutions. The authors also indicate that after keeping in contact with natural schwertmannite with acid water in the laboratory, schwertmannite transformed into goethite plus hydronium jarosite and later transformed into goethite. After the processes described, more than 99% As and Pb remained in the solid phase throughout the entire aging process, Acero et al., 2006 [24]. Similar behavior can be observed in weathering zones of sulfide deposits, mainly of galena, and in acidic-sulfate-rich environments (acid mine drainage). In this case, plumbojarosite and lead–arsenic jarosite can be formed, Smith et al. 2006 [25].
Ochreous precipitates generated from acid sulfate waters in a mine of the Rosia Montana Gold (Romania) are characterized by high concentrations of Cr, Co, Ni, Cu, Zn, As, Cd, and Pb. A mixture of jarosite and schwertmannite was formed as stable secondary minerals along the investigated transect of the Roşia River. The comparative analysis of the precipitates and waters of the Roşia Montană mine indicates that pH–Eh oscillations would cause mineralogical transformations that could lead to trace elements mobilization in the environment, Azzali et al., 2014 [26].
In the Sarcheshmeh copper mine (Iran), waters were of neutral mine drainage (NMD) with high Mn content. Acid mine drainage (AMD) associated with tailings impoundments exhibited high acidity, sulfates, nitrates and chlorides, among elements such as Al, Cd, Cu, Fe and As. The AMD generated at the tailings site is related to sulfide mineral oxidation, especially pyrite and chalcopyrite. Phases such as jarosite, schwertmannite, goethite and ferrihydrite were detected. Among Al-bearing secondary minerals, gibbsite and alunite were also detected in this copper mine. The iron and aluminum oxy-hydroxysulfates and manganesum oxide and hydroxides play an important role in controlling the mobility of metals, Malakooti et al., 2015 [27].
Liu et al., 2016 [28], investigated methods for remediating As-rich AMD solutions by Mn-coated bone char samples with different Mn concentrations, in the absence of jarosite precipitates; there was no jarosite formation in the AMD. Results obtained show that Mn-coated bone char has up to 78 times more adsorption capacity for As(V) than uncoated bone char. The retention intensity of As(V) increases with the degree of Mn coating on the bone char.
Zhou et al., 2017 [29], studied the removal of contaminants contained in acid mine drainage. Concretely, the authors investigated the effect of biologically formed nano-sized FeS-coated limestone on the retention of As(V), Sb(V) and Cr(VI) contained in AMD. Fe-reducing bacteria and sulfate-reducing bacteria were used for generating these coatings. The authors suggested that the FeS formed is effective for removing the indicated elements from the solution.
A summary of papers from different researchers related to the genesis of jarosite materials is included in Table 2.
Table 2.
Summary of works about natural jarosite formation.
2.2. Synthetic Jarosites
Jarosite-type compounds have been synthesized in the laboratory (as biotic and abiotic procedures), and at an industrial scale. Laboratory syntheses were carried out to determine the structure, thermodynamic parameters, and solubility products of the end member. Syntheses were also carried out to determine the reactivity of jarosite-type compounds for metal recovery, such as gold, silver, copper, and so on, and to investigate the extent of incorporation of lead, and arsenic, among others, for immobilization purposes. At the industrial scale, jarosites are precipitated to remove iron and control sulfates, essentially in zinc hydrometallurgy.
2.2.1. Chemical Synthesis of Jarosite
One of the first researchers to synthesize jarosite and alunite was the german Mitscherlich [30] in 1861. Fairchild, 1933 [31], synthesized potassium, rubidium, lead and silver jarosites by heating the corresponding sulfate solutions at temperatures between 110 and 200 °C. This author indicates that it is possible to separate potassium from cesium by this method since cesium jarosite cannot be synthesized.
Dutrizac and Kaiman, 1976 [1], synthesized jarosites of hydronium, sodium, potassium, rubidium, silver, ammonium, thallium, lead and mercury. Sodium jarosite and ammonium jarosite were prepared from a solution containing sodium (or ammonium) and iron (III) sulfate and sulfuric acid at 95 °C. Potassium, rubidium and thallium jarosites were synthesized from a solution containing the corresponding nitrates, iron (III) sulfate and sulfuric acid. Hydronium and silver jarosites were prepared in an autoclave at 140 °C, using iron (III) sulfate solution and without sulfuric acid (for hydronium jarosite), and silver sulfate and sulfuric acid for silver jarosite. Lead and mercury jarosites were prepared from an iron (III) sulfate solution with sulfuric acid and added slowly to the corresponding solutions of lead nitrate or mercury nitrate at 95 °C. The syntheses elapsed between one-and-a-half hours and three hours of the process. A deficiency in the “alkali” position and an excess of water appeared. This deficiency is due to a hydronium substitution in the alkali metal position.
Additional information about the synthesis of (Na,K,Rb,NH4) jarosites appears in the paper of Dutrizac, 1983 [32]. The most significant factors were temperature and retention time. Increasing temperature (70–100 °C) or time (at 97 °C), increased the jarosite production. The composition of precipitates was independent of the physical factors; the alkali content appeared to increase very slightly with increasing temperatures to about 130 °C, and with prolonged retention time.
In the synthesis of hydronium-potassium jarosite, the experiments were developed using a mixture of iron (III) and potassium sulfates; the temperature was 70 °C for 3 h. The experimental conditions were the best to obtain jarosite compounds with reduced particle size. These authors indicate that an increase in the pH from 0.8 to 2.1 decreased the particle size from 3 µm to 200 nm. An increase in the aging time from 0, 3, and 7 days resulted in semispherical, spherical, and euhedral jarosite structures, respectively.
The Rietveld analysis confirmed that the amount of hydronium substitution by potassium increased with an increase in pH, Hernández-Lazcano et al., 2021 [33]. The modified jarosite microstructures can be used as anode materials for improving the lifetime of lithium-ion batteries.
Smith et al., 2006 [11] synthesized jarosite using KOH and Fe2(SO4)3·5H2O. The synthesis was carried out at 95 °C at atmospheric pressure for 4 h. The objective of this synthesis was to study the reactivity of this jarosite at different pH.
Cruells et al., 2000 [34], synthesized a silver potassium jarosite using iron (III), potassium and silver sulfates, and sulfuric acid. The synthesis was carried out at 97 °C for 24 h. Seeding was used to increase the particle size. The atomic relation in the “alkaline site” was: 0.91 potassium; 0.007 silver and 0.083 hydronium. Silver was introduced into the jarosite to determine its cyanidation kinetics.
A study of seeding on the rate of precipitation of ammonium jarosite and sodium jarosite was carried out by Dutrizac, 1996 [35]. Syntheses dissolutions contained iron (III), ammonium sulfates or iron (III) and sodium sulfates. The pH of synthesis was 1.6 (for ammonium) and 1.5 (for sodium). The temperature was 98 °C for a 5–6 h synthesis. Jarosite seed additions significantly increased the rate of jarosite precipitation. Another study carried out by Dutrizac, 2010 [36] compared the rate of precipitation of ammonium jarosite and sodium jarosite. The rate for NH4 jarosite was nine times greater than that for Na-jarosite. The presence of Zn(II) in the precipitation liquids increased the difference.
Patiño et al., 1998 [37], precipitated sodium jarosite using iron (III), sodium and silver sulfates, and sulfuric acid at 97 °C for 24 h. Seeding was used to increase the particle size. The atomic relation in the “alkaline site” was: 0.675 sodium; 0.32 hydronium; and 0.005 silver. Synthesis of sodium jarosite was carried out to develop a study of alkaline decomposition and cyanidation kinetics on the sodium jarosite.
Dutrizac and Chen, 2009 [38], studied the precipitation of potassium jarosite and sodium jarosite in the presence of scandium, yttrium or uranium. A solution containing appropriate concentrations of Fe(SO4)1.5 and K2SO4 or Na2SO4 was prepared, and various amounts of Sc(SO4)1.5, Y(SO4)1.5 or UO2SO4 were added. The precipitation experiments were carried out at 98 ± 0.5 °C. A 24 h or 5 h retention time was employed. Scandium was incorporated in some extension in the Fe position, yttrium was incorporated in small amounts, and uranium was not significantly incorporated.
Dutrizac, 2008 [39], obtained potassium jarosite at 140 °C in chloride media. The production of jarosite, as well as its composition, was independent of the chloride concentration. A temperature of 200 °C increased the jarosite production, but the temperature had no effect on the potassium jarosite composition.
Silver jarosite was synthesized from a dissolution of iron (III) and silver sulfates and sulfuric acid at 97 °C, 24 h. The seed was used to obtain silver jarosite aggregates with sizes up to 100 μm. This synthesis was carried out to determine the behaviour of this jarosite with alkaline decomposition and cyanidation for silver recovery [40].
Smith et al., 2006 [25], synthesized Pb-jarosite using the methods of Dutrizac and Kaiman, 1976 [1] previously described, and Dutrizac et al., 1980 [41]. Lead-arsenic jarosite was developed according to the method described by Alcobé et al., 2001 [42], starting the synthesis process with a dissolution of iron (III) sulfate and arsenic acid. Syntheses were carried out at 95 °C. When the solution temperatures reached 95 °C, a lead nitrate solution was added with stirring. This process was carried out for 5 h. The syntheses were carried out to study the reactivity of this jarosite at different pH.
Islas et al., 2013 [43], synthesized beudantite, as a part of a study on the alkaline reactivity of jarosite-type compounds. The best experimental conditions were: 0.25 M Fe2(SO4)3, 0.02 M H3AsO4, 0.15 M Pb(NO3)2, 94 °C, for 32 h reaction time. The approximate formula of the synthesized sample obtained is as follows: [Pb0.39(H3O)0.22]Fe2.89[(SO4)1.84(AsO4)0.16][(OH)5.51(H2O)0.49]. The beudantite sample is predominantly spherical with a particle size of 30–35 µm. It is made of rhombohedral microcrystals that are strongly bound in a compact structure. This work is part of a wider study on the alkaline reactivity of jarosite-type compounds, which contain arsenic, for determining the conditions for the inactivation of arsenic in these compounds.
2.2.2. Biological Synthesis of Jarosite
During bioleaching of sulfide ores, biooxidation of iron leads to precipitation of jarosite. Jarosite decreases copper bioleaching efficiency. Eftekhari and Kargar, 2018 [44] used Acidithiobacillus ferrooxidans during the synthesis of jarosite. Microorganisms were cultivated in 9K medium containing ferrous, magnesium, and ammonium sulfates, hydrogen potassium phosphate, sodium chloride, and calcium nitrate. These studies were focused on obtaining the minimum precipitation of jarosite during the bioleaching process.
Ammonium jarosite was obtained by microbiological oxidation of ferrous sulfate at pH 2.0–3.0 at 22–65 °C (biotic system). This jarosite was also obtained by chemical procedure (abiotic system) for comparative purposes. In the first method, iron-oxidizing acidophilic microorganisms were used. Schwertmannite was formed at a low amount of ammonium concentration in the synthesis medium, and ammonium jarosite was obtained with a high amount of ammonium at a pH of 1.9- and 7-days incubation. Abiotic jarosites were prepared with ferric sulfate and ammonium sulfate at 36 °C (27 days), 65 °C (8 days), and 95 °C (24 h). The main conclusion of the authors was that the role of microorganisms in the formation of the jarosite includes the catalysis of iron oxidation at low pH and indicates that the formation of mineral species under hyperacid conditions is controlled by geochemical factors, Wang et al., 2007 [45].
Bigham et al., 2010 [46], synthesized jarosite by chemical synthesis. The precipitation solution contained a high level of K+ and iron (III) sulfate; the rest of the solution composition simulated a nutrient medium for bioleaching and iron oxidation (Mg, Ca, nitrates and phosphates in a sulfate matrix). The synthesis temperature was between 2–40 °C for 10 days. All samples contained hydronium and potassium jarosites, being the first majority. The study offers insight into managing Fe(III) and SO4 precipitation at ambient temperature ranges in bioleaching operations and in the development of geochemical models that describe the solubility, speciation, and phase equilibria of iron in sulfate-rich acid leaching systems.
Bigham et al., 2013 [47], developed the precipitation of jarosite in two steps. In the first step, Acidithiobacillus ferroxidans cultures participated in the oxidation of iron (II) at pH 2.4, 8 days. In the second step, lead nitrate was added, and the precipitation was carried out for 22 days. The final precipitate was formed by anglesite, schwertmannite and hydronium jarosite. No lead jarosite was formed.
Jones et al., 2014 [48], synthesized a series of solid solution jarosites by oxidation of ferrous iron at pH 2.2–4.4 and ambient temperature in media containing mixtures of K+ and NH4 solutions. Acidithiobacillus ferrooxidans microorganisms were also used jointly with FeSO4 at pH 2.2 and 22 °C. At low cation concentration, schwertmannite was obtained. With increasing concentrations of K+ and/or NH4+ in the medium, ternary solid solutions of jarosite phases (including hydronium) were obtained. These authors indicate that these solid solutions have the capacity to retain various cations and oxyanions. Thus, their formation is interesting for scavenging impurities in metallurgical process circuits and in impacted natural environments.
Crabbe et al., 2015 [49], studied the formation of jarosite in the presence of amino acids at pH 1.75 and 2.9. These authors indicate that understanding the formation and stability of jarosite in acid environments and in the presence of microbes is of fundamental interest. The presence of amino acid in the precipitation of jarosite reduced the yield and the particle size (at pH 1.75). At higher pH, the impact of amino acids is significantly diminished. The amino acid with a major impact was glycine. Nucleation and growth of jarosite are inhibited in the presence of glycine.
2.2.3. Synthesis of Jarosite in Hydrometallurgical Circuits
Jarosite precipitation is an essential method for control of iron, sulfate and alkali metal ions in hydrometallurgical processing solutions, mainly in the extractive metallurgy of zinc.
In the extraction of this metal, the main process starts with sphalerite roasting, obtaining calcine formed by zinc oxide and zinc ferrite. Solids obtained are leached in two steps with sulfuric acid solutions. Iron contained in the sphalerite concentrate appears in the leaching liquid, jointly with zinc and elements such as lead, copper, indium, gallium, arsenic and cadmium, among others. Potassium and sodium are also present, but due to their low concentration, ammonium—a low-cost reagent—is added to remove the iron from the solution by jarosite precipitation. The resulting liquids are treated with zinc powder to remove elements such as Cd, Cu, etc. Zinc is recovered by electrolysis [50].
To develop the jarosite precipitation, the necessary ions are added to the hot leach liquors, and pH is adjusted with the addition of calcine. Precipitation is carried out at 95 °C, with an acidity of ≈10 g/L, and normally 6 h retention time. The precipitate settles rapidly and is filtered, and after washing, is commonly stored. The jarosite process is applicable to other hydrometallurgical circuits [13,50].
Significant amounts of other elements, such as silver, zinc, cadmium, copper and others, can be incorporated into the jarosite precipitate in a solid solution, and the losses of such metals may be significant from an economic point of view. Numerous authors have developed processes for the recovery of some of these elements, as can be observed in the works of Han et al. 2014 [51]; Kerolli-Mustafa et al., 2016 [52]; Antrekowitsch, 2016 [53]; Steinlechner, 2017 [54]; Wu et al., 2020 [55]).
2.2.4. Synthesis of Alunite
Alunite can be synthesized by the hydrolysis of alum, AlΚ(SO4)2·12H2O, at 12 bar pressure, at a temperature between 175 and 180 °C, and a synthesis time of ≈2 h. A second sample of alunite was synthesized using Al2(SO4)3·18H2O and Κ2SO4 solution and refluxed for 3 h. Τhe pH value of the refluxed solution was 3.0–3.6 [56].
Sunyer and Viñals, 2011 [7] studied the synthesis of natroalunite with the objective of achieving the arsenate substitution in the alunite structure for arsenic immobilization. These authors used numerous experimental conditions of synthesis. Aluminium and sodium sulfates were added to the synthesis medium jointly with variable amounts of arsenate, and in some cases, calcium sulfate. The best temperatures were higher than 160 °C and the retention time was 2 h. The pH varied between 1.0 and 2.8.
The substitution of arsenate for sulfate in natroalunite has also been investigated by Luo et al., 2015 [57]. This study was developed for arsenic immobilization/stabilization. The best conditions for synthesis were pH 3.00 and 200 °C for 3 h. The maximum arsenate substitution observed in the natroalunite structure was approximately 11% molar.
During the acid-leaching process of black shale, a large amount of aluminum was leached. To separate aluminum from the vanadium, the precipitation of alunite and natroalunite was investigated. The synthesis was at 200 °C, pH of 0.4, 5 h. The alunite forms more easily than natroalunite [58].
Zhu et al., 2019 [59], studied the hydrothermal synthesis with a stoichiometric Na:Al:SO4 mole ratio of 1:3:2, using aluminium sulfate and sodium sulfate. Arsenic was incorporated as arsenic acid. The liquids were regulated to pH 4.00 and heated at 200 °C for 2–48 h in a 200 mL stainless steel autoclave. After cooling, the obtained products were filtered, washed three times with pure water, and dried at 110 °C for 24 h. Solubility studies of synthesized natroalunite were carried out. Table 3 shows a summary of experimental conditions for the synthesis of different jarosite materials.
Table 3.
Summary of experimental conditions of synthesis of jarosites.
3. Structure and Thermodynamics of Jarosite–Beudantite–Alunite Solids
Different authors studied the structure and thermodynamic properties of natural and synthetic jarosite, alunite and beudantite.
3.1. Structure of Jarosite-Type Compounds
Cloutis et al., 2006 [60], indicated that the structure of different jarosites: jarosite, natrojarosite, hydronium jarosite and alunite are based on octahedral–tetrahedral sheets. The corner-sharing octahedra form a six-membered ring. At the junction of three six-membered rings is a three-membered ring, and one set of the apical vertices of those three octahedral links to a tetrahedron. The sheets are held together by interstitial cations and hydrogen bonds.
Aguilar-Carrillo et al., 2018 [61], synthesized K, Na, and Pb jarosites and the same compounds with arsenic in their structure. They indicate it seems that when Pb and As incorporate into the structure, they generate an enlargement of the c-axis parameter, as arsenate is larger than sulfate; this can be compensated by the incorporation of Na or Pb with smaller ionic radii than K+ or H3O+. The authors conclude that jarosites have synergistic capacities for incorporating Pb and As into the structure. The simultaneous incorporation of As and Pb confers the jarosites’ lower solubility, immobilizing both ions more efficiently.
Smith et al., 2006 [11], established the structure of synthetic potassium jarosite (with hydronium). These authors indicate that the structure of jarosite is also based on the same linear tetrahedral-octahedral-tetrahedral (T-O-T) sheets, comprised of slightly distorted FeO6 octahedra and sulfate tetrahedra. The calculated lattice parameters of the synthetic jarosite were a0 = 7.3137(6) and c0 = 17.0730(5). The molar ratio K/H3O+ in the formula was 0.84/0.16.
Smith et al., 2006 [25], studied the structure of synthesized Pb-jarosite and Pb-As-jarosite. The structure of these jarosites is as previously described by the same authors for jarosite; the only difference is the partial substitution of sulfate by arsenate in the tetrahedra of the structure. The lattice parameters of Pb-jarosite (in Å) were a0 = 7.3347(7) and c0 = 16.9700(5), using a single, rather than a doubled, unit cell. The lattice parameters for the Pb-As jarosite (in Å), were a0 = 7.3417(1) and c0 = 16.9213(8); the formula obtained were:
(H3O)0.74Pb0.13Fe2.92(SO4)2(OH)5.76(H2O)0.24 for Pb-jarosite.
(H3O)0.68Pb0.32Fe2.86(SO4)1.69(AsO4)0.31(OH)5.59(H2O)0.41, for Pb-As-jarosite.
Ammonium jarosite was produced by microbiological oxidation of ferrous sulfate at pH 2.0–3.0 and 22–65 °C, using thermoacidophiles. Ammonium jarosite was also produced by chemical procedures at 36–95 °C. All samples were deficient in nitrogen compared to stoichiometric ammonium jarosite. Well crystalline materials were obtained with average values of cell parameters c0 = 17.467 ± 0.048 Å, and a0 = 7.330 ± 0.006 Å. A partial replacement of ammonium by hydronium to form solid solutions with 0.14–0.24 mol hydronium per formula unit was detected [45].
Szymanski, 1988 [62], indicates that the structure of beudantite follows the alunite–jarosite model. The structure of beudantite is centrosymmetric, rhombohedral (hexagonal axes), R-3m, and the lattice parameters obtained (in Å) were a = 7.3151(9) and c = 17.0355(5). The lead cation is disordered, and its position is about 0.28 Å, from the origin along x.
According to Frost et al., 2006 [63], the jarosite mineral group can be characterized by its Raman spectra. The spectra of the natural minerals may not be the same as the corresponding synthesized solids. These spectra can be complex. This complexity is attributed to the incorporation of low levels of other cations into the structure in the “alkaline site” or in the central ion position.
The elastic properties of jarosite were investigated by Xu et al., 2010 [64] using synchrotron X-ray diffraction coupled with a multi-anvil apparatus. When pressure over the jarosite sample increased, the parameter c contracted much more rapidly than parameter a, resulting in a large anisotropy in compression. The authors indicated that this behavior is consistent with the layered nature of the jarosite structure, in which the (001) [Fe(O,OH)6]/[SO4] sheets are held together via relatively weak potassium–oxygen and hydrogen bonds.
Sunyer and Viñals, 2011 [65] showed a scheme of the natroalunite (see Figure 2; the structure of jarosites is equivalent, changing Al(III) by Fe(III)). The dashed lines indicate the unit cell, octahedra are formed by Al-O2(OH)4 groups, and the spheres are sodium. The corresponding structural type is R-3m Z = 3, typical of alunite phases. These authors determined the unit cell parameters of arsenical natroalunite, indicating that the a0 values varied between 6.9964 and 7.0058 Å (practically independent of the arsenate and hydronium substitution), and c0 varied between 16.7400 and 16.8149 Å (this parameter increased with arsenate and hydronium substitution).
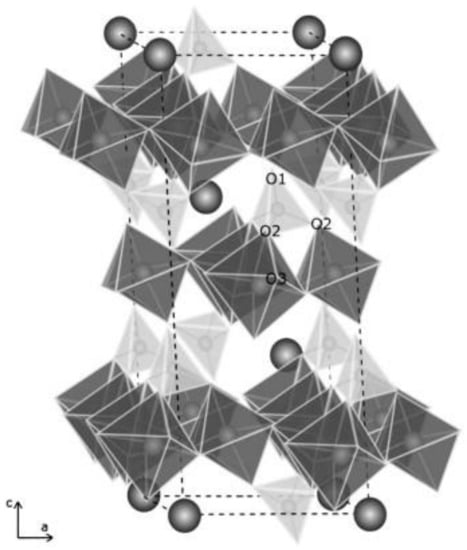
Figure 2.
Scheme of the natroalunite structure, equivalent to the jarosite structure [65].
The structure of alunites is trigonal with a = 6.990Å, c = 16.905Å, with space group R3m, and Z = 3 (Frost and Wain, 2008) [66]. These authors have determined the thermal stability of alunites and indicated that dehydration occurs up to 225 °C, dehydroxylation at 520 °C, and desulphation takes place as a series of steps at 649, 685 and 744 °C.
Drouet and Navrotsky, 2003 [67], determined the unit cell parameters for some synthesized jarosites and alunites. Table 4 shows the values of these parameters for several jarosite–alunite solid solutions. These authors indicate that for jarosite–alunite and sodium jarosite–sodium alunite, the parameter a increases when iron increases. For alunite–sodium alunite, the parameter a is nearly constant, and the parameter c increases with the sodium content.
Table 4.
Unit cell parameters of several samples of jarosite-alunite [66].
3.2. Thermodynamics of Jarosites
A thermochemistry study of jarosite–alunite was carried out by Drouet and Navrotsky [67]. Values of enthalpies of formation of these kinds of solids were determined. Enthalpies of formation for jarosite—alunite solid solutions were between −4778 kJ/mol (Sample 1) and −3963 kJ/mol (Sample 2); these values increase with the potassium content. For natrojarosite—natroalunite solid solutions, values obtained were between −3760 (Sample 3) and −4980 kJ/mol (Sample 4). For alunite–natroalunite solid solutions, values obtained were between −4996 kJ/mol (Sample 5) and −5023 kJ/mol (Sample 6). The values in parentheses correspond to the samples indicated in Table 3.
Perez-Labra et al., 2012 [68], determined some thermodynamic properties of rubidium jarosite. According to their calculus, values of formation obtained were (at 298.15 K): ΔHf = −3806 kJ/mol; ΔSf = −1666 kJ/mol·K; and ΔGf = −3310 kJ/mol. These authors also indicate that rubidium jarosite is stable at pH < 4.7. When this parameter is >4.7, goethite was formed.
The enthalpy of formation of a synthetic Pb-jarosite sample, with the chemical formula (H3O)0.74Pb0.13Fe2.92(SO4)2(OH)5.76(H2O)0.24 was measured by Forray et al., 2010 [69]. Values obtained were: ΔG0f = –3118.1 kJ/mol; S0 = 376.6 J/(mol K) and ΔH0f = −3603.6 kJ/mol. These data can be used for the calculation of phase diagrams of the Pb–Fe–SO4–H2O system and for estimating the solubility product of pure lead jarosite.
The enthalpy of formation for different jarosites, according to Forray et al., 2014 [70], are as follows: (a) for a jarosite Pb-As, with a molar ratio H3O = 0.68/Pb = 0.32; sulfate = 1.69/arsenate 0.31 is = −3691 kJ/mol. (b) for a jarosite Pb-Cu, with a molar ratio H3O = 0.67/Pb = 0.33; Fe = 2.71/Cu 0.25 is −3654 kJ/mol. (c) for a jarosite Pb-Zn, with a molar ratio H3O = 0.57/Pb = 0.43; Fe = 2.70/Zn 0.21 is −3669 kJ/mol. The standard Gibbs free energy of formation from elements at 298 K, were calculated and values obtained were −3165 kJ/mol, −3131 kJ/mol, and −3153.6 kJ/mol, respectively. These data should also prove helpful for the calculation of phase diagrams of different systems.
Zhu et al., 2019 [59], determined under the condition of initial pH 2.00 and 25 °C, the solubility product and the Gibbs free energies of formation for natroalunite. Values obtained were ≈1081 and −4714 kJ/mol, respectively, for natroalunite. For the arsenic-incorporated natroalunite, the corresponding values were ≈1092 and −5078 kJ/mol, respectively. Results obtained indicate that the solubility decreased when arsenic was incorporated into the alunite structure.
4. Reactivity of Jarosites
4.1. Reactivity in Alkaline Media
Many papers studied the decomposition of jarosite–beudantite solids in alkaline media; some of them are kinetic studies of these kinds of reactions. Table 5 shows the kinetic parameter values of alkaline decomposition and, in some cases, cyanidation of synthetic and natural jarosites [16,34,37,40,71,72,73,74,75,76,77,78,79,80,81]. Almost all the processes present an induction period followed by a progressive conversion period until the reaction finishes. The induction period decreases as concentration and temperature increase. The conversion step is characterized by the removal of sulfates and ions corresponding to the “alkaline site”, such as Na, K, ammonium, or hydronium. In all cases, the alkaline decomposition is chemically controlled (see the activation energy values shown in Table 5).
Table 5.
Kinetic parameters of alkaline decomposition of some jarosites.
Cyanidation of jarosites in alkaline media, for silver recovery included in the jarosite structures, consists of two steps in series: (1) alkaline decomposition that controls the kinetics of the process and (2) complexation of silver from the decomposition products. In almost all cases, the kinetics of the cyanidation process is independent of the initial cyanide concentration, except for industrial ammonium jarosite generated in Zn hydrometallurgy, in which the reaction order with respect to cyanide concentration was 0.15.
Viñals et al., 1991 [82], studied the gold and silver recovery from hematite tailings containing plumbojarosite. The first step of the process consisted of a pretreatment with calcium carbonate (up to pH 5–5.5); a second pretreatment with lime at 60 °C, pH 11–12, and, finally, solids obtained were cyanided at 60 °C. Gold and silver extractions were 85–90% and 90–95%, respectively.
González-Ibarra et al., 2006 [83], studied the alkaline decomposition (at 60 °C) of industrial sodium jarosite from zinc hydrometallurgy. This residue contains 150 g Ag/t. These authors conclude that the kinetic of the process in NaOH media is controlled by the chemical reaction, whereas the kinetics of the process in Ca(OH)2 media was controlled through a layer of solid products; this is due to the formation of a calcium–ferrite layer in the surface of the jarosite particles.
Celep and Serbest, 2015 [84], studied the cyanidation of gold and silver in a refractory gossan ore. By mineralogical characterization, gold was detected as native and as electrum. This gold-silver alloy was associated with beudantite and iron oxides. Silver was also present as acanthite and inclusions within beudantite. The extraction level by leaching after 24 h were 76% gold and only 23% silver. The authors indicate that the noble metals are associated with jarosite–beudantite phases. With an alkaline treatment (KOH solutions), prior to cyanidation, the extraction level was 90% Au and 87% Ag.
Table 6 includes values of experimental rate constants (kexp) of cyanidation in NaOH medium (at 50 °C) and in Ca(OH)2 medium (at 50 °C) for several jarosites, corrected for the size factor. Higher rates were obtained by synthetic jarosite in both media. After jarosite appears silver jarosite and ammonium jarosite (in NaOH media), and sodium jarosite and ammonium jarosite in (Ca(OH)2 media). For industrial ammonium jarosite, arsenical jarosite and beudantite, the rates are negligible or almost negligible (at 50 °C). In summary, the cyanidation rate decreases when the substitution level in the jarosite increases, for example, arsenate and/or lead. This fact can be used for the immobilization of lead and arsenic contained in effluents [72,73].
Table 6.
Cyanidation rate of jarosites in NaOH and Ca(OH)2 media [72,73].
4.2. Reactivity in Acid Media
As has been indicated in item 2, jarosite can be generated from the weathering zones of sulphide deposits and acid rock or acid mine drainage. At pH 2 (HClO4), dissolution yields only aqueous products. However, at pH 8, nanoparticles of secondary goethite on the jarosite grain surfaces were formed. These coatings inhibit subsequent jarosite dissolution. All of these results explain the cycling of SO4 and potassium in acid rock and mine drainages. Results obtained confirm the importance of goethite in sulfate environments [11].
Lead– and Pb–As-jarosites are also generated in the same environments and form on or near galena. These solids can be generated from weathering zones of sulphide deposits and acid rock or acid mine drainage. Pb-jarosite at pH 2 (HClO4) dissolves sulfates, lead, and iron. This jarosite at pH 8 (lime) generates lead and sulfates in solution and a nearly amorphous iron hydroxide (III). Arsenical Pb-jarosite at pH 2 (HClO4) generates a poorly crystalline lead sulfate and this compound and iron hydroxide (III) at pH 8. These formed solids are dissolution inhibitors. Arsenate dissolved during leaching or arsenical Pb-jarosite [25].
The dissolution kinetics of an argentian plumbojarosite from the old tailings of sulfating pyrites (Tharsis, Huelva, Spain) for silver recovery at 60–90 °C was studied by Viñals and Núñez, 1988 [85]. The media used was hydrochloric acid–calcium chloride solutions. The activation energy of 96 kJ/mol and a first-order dependence with respect to the hydrochloric acid activity with or without the addition of calcium chloride were obtained. The authors indicate that once plumbojarosite is decomposed, gold can be dissolved by the injection of small amounts of chlorine.
Kendall et al., 2013 [86], studied the dissolution rate of arsenical jarosite in acid media (pH 2), and in water and alkaline media (pH 8). The most important result indicates that the presence of arsenic in the jarosite structure resulted in an increase in dissolution rates in open systems. However, in closed systems, the formation of surface arsenate-iron complexes inhibits further dissolution.
Zhu et al., 2019 [59], revised the dissolution of natroalunite and arsenical natroalunite in acid media (HNO3), at 25–45 °C and pH between 2 and 4. Dissolution in H3O+ medium was near-stoichiometric within the short beginning period, and the dissolved ions were stoichiometric according to the initial solids. Ions were released from solid to solution preferentially through the following order: sodium, hydronium, sulfate, arsenate and aluminium.
Islas et al., 2020 [87], studied the dissolution process of hazardous jarosite-containing toxic elements (Thallium, mercury, lead and lead–arsenic jarosites, all of them with partial substitution of the metal by hydronium). The dissolution behavior of all jarosite types was studied in H3O+ and OH− media. Jarosites are more stable in an acidic medium than in an alkaline medium. The main effects in the jarosite stability were temperature and pH. In this study, the shrinking core kinetic model was applied to understand the stability of hazardous jarosites.
Calla-Choque and Lapidus, 2021 [88] studied the leaching kinetics of an industrial natrojarosite from a Zn leaching plant using thiourea and oxalate in acid media. The temperatures studied were between 25–60 °C and pH between 1 and 3. During leaching, silver contained in the jarosite structure was complexed by thiourea and iron by oxalate. The best conditions were 50 °C and pH 1. The activation energy obtained was 43–44 kJ/mol.
Castro et al., 2017 [89] studied the bioreduction of jarosites; Fe(III)-reducing bacteria (Shewanellaputrefaciens), pH 6.5–7, were used. In 7 days, a maximum reduction of 70% iron was observed. These authors indicate that one key factor for the implementation of anaerobic bioleaching is the metal recovery from the jarosites.
The reduction of different jarosites by a natural bacterial/microbial consortium and particularly, Desulfosporosinus orientis bacteria, was studied [90]. The generation of Fe(II) and the presence of that bacteria are conducted through the precipitation of metal sulphides, immobilizing toxic heavy metals. According to the authors, this study leads to understanding the processes related to the mobility or retention of metals in mine waste and industrial landfill environments.
Dutrizac and Sunyer, 2012 [91] studied the conversion of different jarosite to hematites. For potassium jarosite, conversion was completed in 2 h at 240 °C with seed (hematite). For lead jarosite and sodium–lead jarosite, the process occurred in 0.75 h at 225 °C (with seed). Silver dissolved during the process remained in the final solution.
Sunyer and Viñals, 2011 [65] developed experiments of natroalunite leaching at pH 1 using sulfuric acid. The amount of arsenic solubilized at pH 1 was less than 1 mg/L. These authors indicate that natroalunite is very stable at this acidity level.
4.3. Transformation by Thermal Treatments
Frost et al., 2005 [92] studied the thermal decomposition of natural and synthetic plumbojarosite. These authors indicate the importance of plumbojarosite and argentianplumbojarosite in ancient metallurgy (pre- and Roman times, Colonial Spain in America, and so on). In the thermal decomposition of natural plumbojarosite, dehydroxylation was produced at 391 and 418 °C. The loss of sulfates was detected at 531 °C. Finally, at 812 and 759 °C, losses of oxygen were produced. The temperatures of the thermal decomposition of the natural jarosite were less than that for synthetic lead jarosite.
The hydronium jarosite is stable up to around 200 °C (Frost et al., 2006 [93]). Dehydroxylation occurred at 262, 294, 345 and 385 °C. The proton lost at 557 °C was associated with the loss of sulfate. The remainder of the sulfate was lost at 619 °C.
Sodium and ammonium jarosite precipitates were obtained after sulfuric acid leaching of deep-sea nodules (Vu et al., 2014 [94]). Jarosites decomposed in NaOH or ammonia solutions. Impurities such as Mn, Cu and Ni appeared in the final solids, but copper partially dissolved, complexed by ammonia. The decomposition solids were sintered at 400 °C and 750 °C, and well-crystallized hematite was obtained.
Kerolli et al., 2016 [52] studied the thermal decomposition of ammonium jarosite waste from Mitrovica Industrial Park, Kosovo. Other species detected in solid wastes were gypsum, franklinite and quartz. The decomposition started with the release of adsorbed water between 106 and 152 °C. Ammonia generates between 150 and 300 °C, and SO3 was formed between 421 and 550 °C. At 1000 °C, the products formed were a mixture of magnetite and hematite.
Ma et al., 2019 [95], studied the transformation of an ammonium jarosite from zinc hydrometallurgy by a thermal treatment in a reducing atmosphere, at 300 °C, and in the presence of sulfur. These kinds of studies are interesting to obtain information about the jarosite decomposition and the possibility of recovering valuable elements and protecting the environment. The jarosite decomposed at 300 °C with the formation of szomolnokite (an iron(II) sulfate monohydrate) and iron oxide phases. After thermal treatment at 700 °C, szomolnokite transforms into pyrrhotite and magnetite phases. The activation energy of pyrrhotite formation was determined, obtaining 216 kJ/mol. Pyrrhotite can be used for sulfuric acid production.
Similar work was carried out by the same authors [96] about the thermal decomposition of an ammonium jarosite in the presence of carbon. At <400 °C, the carbon did not affect the decomposition of ammonium jarosite. At >400 °C, the decomposition temperature of the ammonium jarosite with carbon was lower (about 100 °C) than the ammonium jarosite without carbon. Ammonium jarosite decomposed at 400 °C without carbon, forming iron(III) sulfate and intermediate phases. At 700 °C, hematite was the only phase detected. Ammonium jarosite decomposed at 400 °C, with carbon, forming hematite. The activation energy values obtained were 197.7 and 281.4 kJ/mol, respectively.
An alternative for the recovery of metals of interest contained in jarosites was developed by Wang et al., 2019 [97], and Wang et al., 2020 [98]. The basis of the proposed process was a direct reduction of jarosite generated in Zn hydrometallurgy by using coal. Elements such as lead and zinc volatilized during pelletization. Solids obtained were subjected to a magnetic separation. The magnetic fraction contained almost all iron and the non-magnetic fraction was used for removing cadmium, lead, and arsenic from wastewater of Zn hydrometallurgy.
5. Immobilization of Metals in Alunite-Type Structures
Many researchers have devoted great attention to the possibility of immobilization of toxic metals contained in solid and liquid wastes, such as arsenic, lead, mercury, and thallium and so on, in structures of jarosite, beudantite or alunite phases.
Kolitsch and Pring, 2001 [99], determined the suitability of the alunite-type structures, including crandallite (calcium aluminium hydroxilate phosphate), for immobilization of toxic metals in natural and synthesized solids. Examples of elements studied were arsenic, lead, mercury, thallium, chromium and radioactive metals. The authors indicate that the potential in incorporating toxic metals in alunite-type structures is comparable to the same effect in structures such as apatite, perovskite or pyrochlore.
Roussel et al., 2000 [100] characterized solids from the oxidation of sulfide-rich tailings of an old gold mine in La Petite Faye, France. These authors detected the presence of scorodite and plumbojarosite. The solubility of these phases was determined. The authors conclude that scorodite is not a relevant candidate for As-in situ immobilization. The precipitation of beudantite maintained a low lead amount in the solution. Only the presence of lead sulfate generates soluble species, releasing significant amounts of lead into the water.
Smeaton et al., 2012 [101], determined the stability of synthetic arsenic–lead jarosite. These authors demonstrated the reduction of Fe(III) and As(V) during the microbial dissolution (using Shewanella putrefaciens). The presence of bacteria generates a reductive process, and consequently, it makes inappropriate the use of this kind of jarosite for collecting elements such as lead and arsenic.
Alarcón et al., 2014 [102] indicated that compounds of Fe(III) such as jarosite, schwertmannite, goethite and ferrihydrite present efficacy as arsenic sorbents. The authors established the arsenic retention and transport under a marine flood scenario, simulating changes in geochemical conditions in coastal systems due to sea-level rise. Minerals such as those indicated were contacted with arsenic solutions for 25 days. The retention capacity of arsenic was (in wt%): goethite, 3; ferrihydrite, 3; schwertmannite, 0.8 and jarosite, 0.3%.
Aguilar-Carrillo et al., 2018 [61] studied the extent and the effects of simultaneous incorporation of As(V) and Pb(II) into synthetic jarosite at different As/Pb molar ratios. The amount of arsenic during precipitation is larger when lead is incorporated into the structure, but also the amount of lead is larger when arsenic is incorporated. This question provides insight into the relevance of geochemical pathways of both metals immobilization in mining-metallurgical wastes in environments related to acid mine drainage and acid sulfate soils.
Luo et al., 2015 [57] synthesized arsenic-free natroalunite, and natroalunite with arsenic. The maximum amount of arsenate for sulfate was approximately 11% molar. By using amounts of arsenic less than 4 g/L, only natroalunite was obtained. For amounts of As > 4.2 g/L, amorphous aluminium arsenate was formed. Leaching tests over these solids indicate the solubility of arsenic in natroalunite was between 0.1 and 0.8 mg/L in 20 weeks at pH 4–9. However, the same parameter and the same conditions for aluminium arsenate were between 3 and 6.8 mg/L.
Synthetic beudantite and hidalgoite, were synthesized by Zhu et al., 2021 [103]. Studies of leaching were carried out for environmental purposes, indicating that the minimum amount of Pb and As in beudantite were, respectively, 0.0027–0.0030 mg/L (pH 6) and 0.0248–0.0250 mg/L (at pH 4). For hidalgoite, the minimum amount of Pb and As were, respectively, 0.0055–0.0061 mg/L (pH 12) and 0.0750–0.0810 mg/L (at pH 7). Beudantite is more effective for the immobilization of Pb and As than hidalgoite.
The magnitude of arsenic substitution in alunite-type structures (Hudson-Edwards, 2019) [104], (in % weight) is 3–8.5 As in beudantite; 3.6 As in alunite; 2.8 As in natroalunite; 1.6 As in jarosite; 0.034 As in hydroniumalunite. The degree of uptake depends on the AsO4/TO4 ratio. Arsenic release from alunite and natroalunite is limited (pH 5–8 in aerobic and abiotic arsenic). Similar behavior was detected in jarosites at pH 8. The release of arsenic is limited due to the stability of structures or due to the formation of other structures that resorbed the released arsenic.
Viñals et al., 2010 [6] studied the hydrothermal precipitation of As(V) as arsenical natroalunite. Synthesis was carried out at 200 °C, 15 min, using aluminium and sodium sulfates for arsenic inertization, from residues of copper pyrometallurgy. The maximum arsenic substitution in natroalunite was 0.07 in the formula for Al/As = 4.5 (molar ratio). In the solubility of precipitates, the amount of arsenic at pH 1 was 0.5–2 mg/L. Between pH 6 and 10, the amount of arsenic solubilized was near 0.01 mg/L. The precipitation of arsenic into natroalunite is a very good method for arsenic immobilization.
The maximum stability of arsenical natroalunite was between pH 5 to 8, obtaining an arsenic dissolution of 0.01–0.05 mg/L in 24 h (Sunyer and Viñals, 2011 [65]. The dissolution is congruent at pH < 3, incongruent at pH 4–11, and congruent at pH 12. Precipitates containing other phases as alarsite or amorphous aluminium–arsenate indicated arsenic solubilization as two or three orders of magnitude greater. The use of alunite (central ion Al3+) for toxic element immobilization has the advantage over using jarosite or beudantite (central ion Fe3+). These latter structures can decompose in reducing environments (Fe3+ to Fe2+); this does not occur in alunite.
Johnston et al., 2012 [105], studied the sulfidization of synthetic arsenical jarosite at a pH between 4.0 and 8.0. These authors indicate that in acid mine drainage and acid sulfate soils, the presence of sulfate-reducing bacteria can generate sulfide. Abiotic sulfidization transformed arsenical jarosite to mackinavite (an iron sulfide), enhancing the mobility of arsenic. The rate of jarosite dissolution increased from pH 4.0 to pH 8.0. Arsenic dissolved more and faster at pH 4.0–5.0 compared to pH 6.5–8.
6. Jarosite Passivation of Chalcopyrite during Leaching or Bioleaching
The importance of pH in the chemical leaching of chalcopyrite was established by Córdoba et al., 2009 [106], indicating that this parameter is related to controlling the hydrolysis of ferric species. Jarosites formed on the chalcopyrite surface seem to be responsible for chalcopyrite passivation. These authors indicated that sulfur formed is porous and does not contribute to the passivation of chalcopyrite.
Córdoba et al., 2009 [107] studied the same reaction but in the presence of silver ions (catalytic effect). The authors indicate that in some cases, the presence of silver can prevent the formation of jarosite in the chalcopyrite surface. However, in other cases, when jarosite was formed, silver was incorporated into the jarosite structure, losing the catalytic effect of the silver ion in the chalcopyrite leaching.
Kartal et al., 2020 [108] studied the chalcopyrite leaching in the presence of tetrachloroethylene and dimethyl sulfoxide at 75 °C and 750 mV (SHE). At the later leaching step, potassium jarosite covered the undissolved chalcopyrite. These authors indicate that the careful control of a solution’s composition to remove the possible layer of jarosite is necessary.
Quezada et al. studied a process of agglomeration, curing and leaching of chalcopyrite. A chalcopyrite sample especially rich in copper (28.5% Cu), [109], and a copper ore sample with a copper content of 0.79% Cu, [110], were used. Reagents were sulfuric acid and sodium chloride solutions for curing and sulfuric acid and Cu(II) solutions during leaching. The presence of natrojarosite in the curing products and in the leaching products was detected. These authors indicate that elemental sulfur and natrojarosite are responsible for the passivation of chalcopyrite leaching. The passivation in leaching was more important at low temperatures (compact products on the chalcopyrite surface), than at elevated temperatures (porous products).
During bioleaching of chalcopyrite in the presence of Acidithiobacillus ferrooxidans, potassium jarosite was detected before the formation of ammonium jarosite. These authors indicate that these jarosites covered the chalcopyrite surface with a passivating effect. Passivation of chalcopyrite may be reduced by controlling the jarosite precipitation [111].
Vakylabad et al., 2022 [112], studied the influence of sodium chloride addition in the bioleaching process of chalcopyrite. The presence of chloride ions attempts to mitigate the jarosite formation on chalcopyrite, inhibiting the progress of the reaction. Mesophilic and thermophilic microorganisms were used. Working in columns with an oxidation-reduction potential (ORP) < 450 mV reduced the formation of the passivating layer that was essentially formed by jarosite.
7. Jarosites on Mars
Indications of the presence of jarosites, together with other iron oxides such as goethite, lepidocrocite or hematite, were observed through a study carried out by Morris et al., 2000 [113], on samples from Ares Vallis on the surface of Mars. Analyzes were carried out using the spectrophotometers of the Pathfinder space vehicle, launched by NASA in 1996. These studies and others carried out from these dates are based on the idea of detecting the possible presence of water on Mars.
Analysis using the Mösbauer analytical technique was carried out on rocks and minerals at Meridiani Planum on Mars [18,114]. This technique was mounted in the Opportunity rover (MER-B), launched in 2003 as part of the NASA’s Mars Exploration program. Jarosite was detected jointly with hematite, olivine and pyroxene. The jarosite detected there, can be a mineralogical evidence for the presence of water, probably under acid-sulfate conditions.
Cloutis et al., 2006, [60], indicated that optical spectroscopy could be a powerful tool for sulfate detection and discrimination on a number of planetary bodies such as Europa, Io, Mars, and so on. This is based on the spectral diversity of sulfates and the availability of wavelength regions where sulfates exhibit aspects that are not overlapped by absorption bands of rock-forming minerals.
Madden et al., 2006 [115] studied the kinetics of jarosite dissolution in the sulfuric acid system, at pH between 1.6 and 4.5, at 280–313 K. These authors determined the mineralogy of the leaching residues. They indicate that sulfate adsorption may play a significant role in determining the mineralogy and texture of the iron oxides produced. Goethite is commonly found as the dominant iron oxide related to jarosite deposits on Earth; on Mars, under water-limited conditions, leaching generates nanoscale maghemite and hematite. It seems that abundant iron oxides detected at Meridiani Planum may have formed as secondary reaction products of jarosite dissolution at pH > 3.5 over time less than tens to thousands of years.
Characterization studies were carried out by Rampe et al., 2017 [116], on Mount Sharp on Mars, obtained from the equipment of the Curiosity rover launched in 2014. In the Murray formation, phyllosilicates, hematite, jarosite, and pyroxene were the most abundant minerals detected. These authors indicated that these minerals and particularly hematite and jarosite, are products of acid-sulfate alterations on Earth. They also suggested that influxes of acidic pore fluids generated the diagenesis of the Murray formation and its mineralogy and geochemistry. The preservation of minerals such as jarosite, which are highly susceptible to dissolution at low pH, suggests that fluids may not have been extremely acidic (pH > 2).
Potter-McIntyre and McCollom, 2018 [117], determined the origin of jarosite and alunite at Mollies Nipple (Kane County, UT, USA) and demonstrated that they could be more stable than the current knowledge indicates is possible. The idea of this work was to obtain results for an interpretation of Martian depositional and diagenetic environmental conditions. It seems that previous interpretations of Mars diagenetic environments as being hyperacidic or lacking significant contact with fluids during geologic time frames based on the presence of jarosite and/or alunite may be incorrect. These environments may have had much more circumneutral (and therefore more habitable) diagenetic histories than previously imagined.
The sulfate and iron oxide deposits in Rio Tinto, Huelva, Spain, are similar to early Martian hematite-rich regions. The authors report a successful demonstration of autonomous drilling down to 1 m depth as a proof-of-concept study. The multi-analytical approach informed that molecular biomarkers could be recovered from drilling on iron-rich Mars analogues by using an automated life detection lander prototype. This equipment can be used to search for life in the Martian ice-cemented subsurface [118].
Baccolo et al., 2021 [119], studied the jarosite formation in the deep Antarctic Ice. These authors attempt to explain the formation of jarosite on Mars. They indicate that jarosite is a product of weathering involving Aeolian dust and acidic atmospheric aerosols. It seems reasonable that the formation of jarosite on Mars involves the interaction of brines and mineral dust in deep ice.
The Smoking Hills in Arctic Canada have a widespread occurrence of metal-sulfide mineral deposits. High-temperature oxidation processes generated several oxidized species such as jarosite in bands within phyllosilicate mudstone. The authors indicate that Smoking Hill can be an alternative Earth analogue to Mars. Jarosite-rich layers reflect post-depositional oxidation, not acidic conditions, during the deposition process [120].
8. Conclusions
Jarosite belongs to the alunite supergroup with alunite, beudantite and crandallite. Alunite-type compounds are common on the Earth’s surface. Multiple substitutions in different positions of their structure are possible, and compounds obtained have good stability. These minerals play an important role in different fields of extractive metallurgy; their importance appears when viewing the high number of publications found in the literature.
Jarosite–alunite-type compounds occur: (1) as oxidized parts of sulfide deposits; (2) as nodules and dissemination in clays; (3) in acid soils; (4) as hypogene minerals. The weathering process can be originated in situ on deposits of primary and secondary sulfides or at some distance from the original deposits, where these compounds precipitated after hydrolysis reactions.
Jarosite–alunite-type compounds have been synthesized in the laboratory and at the industrial level. Synthesis in the abiotic and biotic procedures was developed to determine structures, thermodynamic parameters, and solubility products of jarosite and alunite. Synthesis was also carried out to acquire information about the reactivity of these kinds of solids for valuable metals recovery and immobilization of elements such as As, Pb, Hg, and Cr, among others. The structure of the alunite-supergroup is rhombohedral, and the structural group is R-3m, with Z = 3. For a selected group of jarosite–beudantite–alunite minerals, parameter a varied between 6.981 and 7.327 nm, and parameter c varied between 16.635 and 17.087 nm.
Many papers refer to the alkaline decomposition of jarosite–beudantite. Almost all the papers indicate that the process consists of an induction period followed by a conversion period. This second period is characterized by the removal of sulfates and ions corresponding to the “alkaline” site. In all cases, the process is chemically controlled.
The cyanidation of different jarosite and beudantite for silver recovery consists of two steps: an alkaline decomposition that controls the kinetics of the process, followed by the complexation of silver from the decomposition products. A comparison of the cyanidation rate process for several jarosites in NaOH and Ca(OH)2 media indicates that higher rates were obtained by synthetic jarosite (potassium jarosite) in both media. Then appeared silver jarosite and ammonium jarosite (NaOH media), and sodium jarosite and ammonium jarosite (Ca(OH)2 media). For industrial ammonium jarosite (Zn hydrometallurgy), natural arsenical jarosite, and natural beudantite, the rates are almost negligible (at 50 °C). The cyanidation rate decreases when the substitution level in the jarosite increase, for example, substitutions by arsenate or lead.
In general, jarosite-type compounds are more stable in an acid medium than in an alkaline medium. However, this type of solids dissolves in an acid medium, at a pH below 2, obtained through the use of sulfuric, nitric, perchloric or hydrochloric acid, sometimes with a mixture of hydrochloric acid and metal chlorides. The reactivity of jarosites in biotic media has also been studied through bioreduction processes and, in some cases, with the formation of metal sulfides, immobilizing toxic elements. The thermal treatment of jarosites has also been studied, in some cases with the transformation of jarosite into hematite, mixtures of magnetite and hematite, or mixtures of magnetite and pyrrhotite.
The alunite-type compounds (alunite, jarosite, beudantite) exhibited good stability in a wide range of environmental conditions; they have been used for the long-term storage of toxic elements. Arsenic–lead jarosites have been proposed for immobilization of lead and arsenic. Alunite structures have also been proposed for the immobilization of arsenic, lead, mercury, thallium, and chromium, among others. The maximum stability of arsenical natroalunite was between pH 5 to 8, obtaining an arsenic dissolution of 0.01–0.05 mg/L in 24 h. The corresponding authors indicate the use of alunite (central ion Al3+) for toxic elements immobilization has an advantage over using jarosite or beudantite (central ion Fe3+). These latter structures can decompose in reducing environments (Fe3+ to Fe2+). This fact does not occur in alunite.
The sulfidization of synthetic arsenical jarosite at a pH between 4.0 and 8.0 is an example of reducing environments. Sulfidization transformed arsenical jarosite to mackinavite (an iron sulfide), enhancing the mobility of arsenic. The rate of jarosite dissolution increased from pH 4.0 to pH 8.0. Arsenic dissolved more and faster at pH 4.0–5.0 compared to pH 6.5–8.
Jarosites formed in the mineral surface during curing and/or leaching or bioleaching of chalcopyrite seem to be responsible for chalcopyrite passivation. The importance of pH in the leaching process of chalcopyrite is related to the control of the hydrolysis of ferric species present in the system. In certain conditions, the sulfur formed during the process was porous and did not contribute to the passivation of chalcopyrite; in other experimental conditions, the passivation process is a contribution of jarosite and elemental sulfur.
Jarosite, alunite, and minerals such as goethite, hematite, maghemite, lepidocrocite, olivine, and pyroxene, among other species, have been detected on Mars from the results obtained in the spectrophotometers incorporated in Pathfinder, Opportunity, and Curiosity space vehicles launched by NASA. Numerous researchers have carried out studies on different regions of the Earth, in which there may be similarities with various areas of Mars. The minerals detected on Mars can be a piece of mineralogical evidence for the presence of water, probably under acid-sulfate conditions.
Author Contributions
Conceptualization, M.C. and A.R.; methodology, A.R. and M.C.; writing—original draft preparation, A.R.; writing—review and editing, M.C.; visualization, M.C. and A.R.; supervision, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work is dedicated to the memory of John E. Dutrizac (CANMET, Canada) and Joan Viñals (Universitat de Barcelona, Spain), who passed away some years ago. They were great experts in the field of Extractive Metallurgy and, in particular, in studies of precipitation, properties and reactivity of minerals of jarosite-type compounds, as can be seen throughout this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dutrizac, J.E.; Kaiman, S. Synthesis and properties of jarosite-type compounds. Can. Mineral. 1976, 14, 151–158. [Google Scholar]
- Jambor, J.L. Nomenclature of the alunite supergroup. Can. Mineral. 1999, 37, 1323–1341. [Google Scholar]
- Botinelly, T. A review of the minerals of the alunite–jarosite, beudantite, and plumbogummite groups. J. Res. US Geol. Surv. 1976, 4, 213–216. [Google Scholar]
- Scott, K.M. Nomenclature of the alunite supergroup: Discussion. Can. Mineral. 2000, 38, 1295–1297. [Google Scholar] [CrossRef]
- Smith, D.K.; Roberts, A.C.; Bayliss, P.; Liebau, F. A systematic approach to general and structure-type formulas for minerals and other inorganic phases. Am. Mineral. 1998, 83, 126–132. [Google Scholar] [CrossRef]
- Viñals, J.; Sunyer, A.; Torres, E.; Beltran, V.; Llorca, N. Arsenic inertization from copper pyrometallurgy through phases of the alunite supergroup. World Metall.-Erzmetall 2010, 63, 310. [Google Scholar]
- Sunyer, A.; Viñals., J. Arsenate substitution in natroalunite: A Potential Medium for Arsenic Immobilization. Part 1: Synthesis and Compositions. Hydrometallurgy 2011, 109, 54–64. [Google Scholar] [CrossRef]
- Eftekhari, N.; Kargar, M.; Zamin, F.R.; Rastakhiz, N.; Manofi, Z. A review on various aspects of jarosite and its utilization potentials. Ann. Chim. Sci. Mater. 2020, 44, 43–52. [Google Scholar] [CrossRef]
- Das, G.K.; Acharya, S.; Anand, S.; Das, R.P. Jarosites: A Review. Miner. Proccess. Extr. Metall. Rev. 1996, 16, 185–210. [Google Scholar] [CrossRef]
- Hoeber, L.; Steinlechner, S. A comprehensive review of processing strategies for iron precipitation, residues from zinc hydrometallurgy. Clean. Eng. Technol. 2021, 4, 100214. [Google Scholar] [CrossRef]
- Smith, A.M.L.; Hudson-Edwards, K.A.; Dubbin, W.E.; Wright, K. Dissolution of jarosite [KFe3(SO4)2(OH)6] at pH 2 and 8: Insights from Batch Experiments and Computational Modelling. Geochim. Cosmochim. Acta 2006, 70, 608–621. [Google Scholar] [CrossRef]
- Hofstra, A.H.; Snee, L.W.; Rye, R.O.; Folger, H.W.; Phinisey, J.D.; Loranger, R.J.; Dahl, A.R.; Naeser, C.W.; Stein, H.J.; Lewchuk, M.T. Age constraints on Jerritt Canyon and other Carlin-type gold deposits in the Western United States: Relationship to mid-Tertiary extension and magmatism. Econ. Geol. 1999, 94, 769–802. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Jambor, J. Jarosites and their application in Hydrometallurgy. Rev. Mineral. Geochem. 2000, 40, 405–452. [Google Scholar] [CrossRef]
- Velasco, F.; Herrero, J.M.; Suárez, S.; Yusta, I.; Alvaro, A.; Tornos, F. Supergene features and evolution of gossans capping massive sulphide deposits in the Iberian Pyrite Belt. Ore Geol. Rev. 2013, 53, 181–203. [Google Scholar] [CrossRef]
- Viñals, J.; Roca, A.; Cruells, M.; Núñez, C. Characterization and cyanidation of Rio Tinto gossan ores. Can. Metall. Q. 1995, 34, 115–122. [Google Scholar] [CrossRef]
- Roca, A.; Viñals, J.; Arranz, M.; Calero, J. Characterization and Alkaline Decomposition/Cyanidation of Beudantite–Jarosite Materials from Rio Tinto Gossan Ores. Can. Metall. Q. 1999, 38, 93–103. [Google Scholar] [CrossRef]
- Savage, K.S.; Bird, D.K.; O’Day, P.A. Arsenic speciation in synthetic jarosite. Chem. Geol. 2005, 215, 473–498. [Google Scholar] [CrossRef]
- West, L.; McGown, D.J.; Onstott, T.C.; Morris, R.V.; Suchecki, P.; Pratt, L.M. High Lake gossan deposit: An Arctic Analogue for Ancient Martian Surficial Processes? Planet. Space Sci. 2009, 57, 1302–1311. [Google Scholar] [CrossRef]
- Dill, H.G.; Melcher, F.; Kaufhold, S.; Techmer, A.; Weber, B.; Bäumler, W. Post-Miocene and Bronze-age supergene Cu-Pb arsenate-humate-oxalate-carbonate mineralization at Mega Livadi, Serifos, Greece. Can. Mineral. 2010, 48, 163–181. [Google Scholar] [CrossRef]
- Reynolds, G.A. The Jarosite Group of Compounds—Stability, Decomposition and Conversion. Master’s Thesis, University of Newcastle, Newcastle, NSW, Australia, 2007. Available online: https://nova.newcastle.edu.au/vital/access/services/Download/uon:743/ATTACHMENT02 (accessed on 24 March 2022).
- Holley, E.A.; Monecke, T.; Bissig, T.; Reynolds, T.J. Evolution of high-level magmatic-hydrothermal systems: New Insights from Ore Paragenesis of the Veladero High-Sulfidation Epithermal Au–Ag Deposit, El Indio-Pascua Belt, Argentina. Econ. Geol. 2017, 112, 1747–1771. [Google Scholar] [CrossRef]
- Cutruneo, C.M.N.L.; Oliveira, M.L.S.; Ward, C.R.; Hower, J.C.; de Brum, I.A.S.; Sampaio, C.H.; Kautzmann, R.M.; Taffarel, S.R.; Teixeira, E.C.; Silva, L.F.O. A mineralogical and geochemical study of three Brazilian coal cleaning rejects: Demonstration of electron beam applications. Int. J. Coal Geol. 2014, 130, 33–52. [Google Scholar] [CrossRef]
- Montero, I.C.; Brimhall, G.H.; Alpers, C.N.; Swayze, G.A. Characterization of waste rock associated with acid drainage at the Penn Mine, California, by ground-based visible to short-wave infrared reflectance spectroscopy assisted by digital mapping. Chem. Geol. 2005, 215, 453–472. [Google Scholar] [CrossRef]
- Acero, P.; Ayora, C.; Torrentó, C.; Nieto, J.M. The behavior of trace elements during schwertmannite precipitation and subsequent transformation into goethite and jarosite. Geochim. Cosmochim. Acta 2006, 70, 4130–4139. [Google Scholar] [CrossRef]
- Smith, A.M.; Dubbin, W.E.; Wright, K.; Hudson-Edwards, K.A. Dissolution of lead- and lead-arsenic-jarosites at pH 2 and 8 and 20 °C: Insights from batch experiments. Chem. Geol. 2006, 229, 344–361. [Google Scholar] [CrossRef]
- Azzali, E.; Marescotti, P.; Frau, F.; Dinelli, E.; Carbone, C.; Capitani, G.; Luchetti, G. Mineralogical and chemical variations of ochreous precipitates from acid sulphate waters/asw) at the Rosia Montana gold mine (Romania). Environ. Earth Sci. 2014, 72, 3567–3584. [Google Scholar] [CrossRef]
- Malakooti, S.J.; Shahhosseini, M.; Ardejani, F.D.; Tonkaboni, S.Z.S.; Noaparast, M. Hydrochemical characterisation of water quality in the Sarcheshmeh copper complex, SE Iran. Environ. Earth Sci. 2015, 74, 3171–3190. [Google Scholar] [CrossRef]
- Liu, J.; He, L.; Dong, F.; Hudson-Edwards, K.A. The role of nano-sized manganese coatings on bone char in removing arsenic (V) from solution: Implications for Permeable Reactive Barrier Technologies. Chemosphere 2016, 153, 146–154. [Google Scholar] [CrossRef]
- Zhou, L.; Dong, F.; Liu, J.; Hudson-Edwards, K.A. Coupling effect of Fe3+(aq) and biological, nano-sized FeS-coated limestone on the removal of redox-sensitive contaminants (As,Sb and Cr): Implications for In Situ Passive Treatment of Acid Mine Drainage. Appl. Geochem. 2017, 80, 102–111. [Google Scholar] [CrossRef]
- Mitscherlich. J. Prakt. Chem. Leipz. 1861, 83, 474. Available online: https://www.mindat.org/min-10725.html (accessed on 24 March 2022).
- Fairchild, J.G. Artificial Jarosites-the separation of potassium from Cesium. Am. Mineral. J. Earth Planet. Mater. 1933, 18, 543–547. Available online: http://www.minsocam.org/ammin/AM18/AM18_543.pdf (accessed on 24 March 2022).
- Dutrizac, J.E. Factors affecting alkali jarosite precipitation. Metall. Trans. B 1983, 14, 531–539. [Google Scholar] [CrossRef]
- Hernández-Lazcano, E.; Cerecedo-Sáenz, E.; Hernández-Ávila, J.; Toro, N.; Karthik, T.V.K.; Mendoza-Anaya, D.; Fernández-García, M.E.; Rodríguez-Lugo, V.; Salinas-Rodríguez, E. Synthesis of Hydronium-Potassium Jarosites: The Effect of pH and Aging Time on Their Structural, Morphological, and Electrical Properties. Minerals 2021, 11, 80. [Google Scholar] [CrossRef]
- Cruells, M.; Roca, A.; Patiño, F.; Salinas, E.; Rivera, I. Cyanidation kinetics of argentian jarosite in alkaline media. Hydrometallurgy 2000, 55, 153–163. [Google Scholar] [CrossRef]
- Dutrizac, J.E. The effect of seeding on the rate of precipitation of ammonium jarosite and sodium jarosite. Hydrometallurgy 1996, 42, 293–312. [Google Scholar] [CrossRef]
- Dutrizac, J.E. Comparative rates of precipitation of ammonium jarosite and sodium jarosite in ferric sulfate-sulfuric acid media. Can. Metall. Q. 2010, 49, 121–130. [Google Scholar] [CrossRef]
- Patiño, F.; Salinas, E.; Cruells, M.; Roca, A. Alkaline decomposition-cyanidation kinetics of argentian natrojarosite. Hydrometallurgy 1998, 49, 323–336. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Chen, T.T. The behaviour of scandium, yttrium and uranium during jarosite precipitation. Hydrometallurgy 2009, 98, 128–135. [Google Scholar] [CrossRef]
- Dutrizac, J.E. Factors affecting the precipitation of potassium jarosite in sulfate and chloride media. Metall. Trans. B 2008, 39, 771–783. [Google Scholar] [CrossRef]
- Roca, A.; Patiño, F.; Viñals, J.; Núñez, C. Alkaline decomposition-cyanidation kinetics of argentojarosite. Hydrometallurgy 1993, 33, 341–357. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Dinardo, O.; Kaiman, S. Factors affecting lead jarosite formation. Hydrometallurgy 1980, 5, 305–324. [Google Scholar] [CrossRef]
- Alcobé, X.; Bassas, J.; Tarruella, I.; Roca, A.; Viñals, J. Structural characterization of synthetic beudantite-type phases by Rietveld refinement. Mater. Sci. Forum 2001, 378, 671–676. [Google Scholar] [CrossRef]
- Islas, H.; Patiño, F.; Flores, M.U.; Reyes, I.A.; Reyes, M.C.M.; Hernández, J. Synthesis and characterization of beudantite. Eur. Metall. Conf. EMC 2013, 1, 303–312. Available online: https://www.researchgate.net/profile/Ivan-Reyes-8/publication/273319348_Synthesis_and_Characterization_of_Beudantite/links/54fe09070cf2741b69ef9a71/Synthesis-and-Characterization-of-Beudantite.pdf (accessed on 24 March 2022).
- Eftekhari, N.; Kargar, M. Assessment of optimal iron concentration in the precipitation of jarosite and the activity of acidithiobacillus ferroxidans. J. Biotechnol. 2018, 9, 525–529. [Google Scholar]
- Wang, H.; Bigham, J.M.; Jones, F.S.; Tuovinen, O.H. Synthesis and properties of ammoniojarosites prepared with iron-oxidizing acidophilic microorganisms at 22–65 °C. Geochim. Cosmochim. Acta 2007, 71, 155–164. [Google Scholar] [CrossRef]
- Bigham, J.M.; Jones, F.S.; Özkaya, B.; Sahinkaya, E.; Puhakka, J.A.; Tuovinen, O.H. Characterization of jarosites produced by chemical synthesis over a temperature gradient from 2 to 40 °C. Int. J. Miner. Proccess. 2010, 94, 121–128. [Google Scholar] [CrossRef]
- Bigham, J.M.; Algur, Ö.F.; Jones, F.S.; Tuovinen, O.H. Solid-phase controls on lead partitioning in laboratory bioleaching solutions. Hydrometallurgy 2013, 136, 27–30. [Google Scholar] [CrossRef]
- Jones, F.S.; Bigham, J.M.; Gramp, J.P.; Tuovinen, O.H. Synthesis and properties of ternary (K,NH4,H3O)-jarosites precipitated from Acidithiobacillus ferrooxidans cultures in simulated bioleaching solutions. Mater. Sci. Eng. C 2014, 44, 391–399. [Google Scholar] [CrossRef]
- Crabbe, H.; Fernandez, N.; Jones, F. Crystallization of jarosite in the presence of amino acids. J. Cryst. Growth 2015, 416, 28–33. [Google Scholar] [CrossRef]
- Sinclair, S.J. The Extractive Metallurgy of Zinc, 1st ed.; Spectrum Series; The Australasian Institute of Mining and Metallurgy: Victoria, Australia, 2005; Volume 13. [Google Scholar]
- Han, H.; Sun, W.; Hu, Y.; Jia, B.; Tang, H. Anglesite and silver recovery from jarosite residues through roasting and sulfidization-flotation in zinc hydrometallurgy. J. Hazard. Mater. 2014, 278, 49–54. [Google Scholar] [CrossRef]
- Kerolli-Mustafa, M.; Mandić, V.; Ćurković, L.; Šipušić, J. Investigation of thermal decomposition of jarosite tailing waste—A prerequisite for comprehensive jarosite reuse and waste minimization. J. Therm. Anal. Calorim. 2016, 123, 421–430. [Google Scholar] [CrossRef]
- Antrekowitsch, J. Recycling of Poly-Metallic Residues from Metal Industry—Current Status and Future Developments. In REWAS 2016; Blanplain, B., Meskers, C., Olivetti, E., Apelian, D., Mishra, B., Howarter, J., Kvithyld, A., Neelameggham, N.R., Spangenberger, J., Eds.; TMS Series; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Steinlechner, S. Present status in the recycling of industrial residues from lead, zinc and copper industry and their possible future contribution to the supply of selected minor elements, like silver. In Proceedings of the METAL 2017—26th International Conference on Metallurgy and Materials, Brno, Czech Republic, 24–26 May 2017; pp. 1469–1474. Available online: http://www.scopus.com/inward/record.url?scp=85043358656&partnerID=8YFLogxK. (accessed on 24 March 2022).
- Wu, J.; Chai, L.; Lin, Z.; Wei, Y.; Shi, M.; Peng, J.; Peng, N.; Yan, X. Fe (II)-induced transformation of Jarosite residues generated from zinc hydrometallurgy: Influence on Metals Behaviors During Acid Washing. Hydrometallurgy 2020, 200, 105523. [Google Scholar] [CrossRef]
- Paszkowicz, W.; Dynowska, E.; Świerkocki, J. X-ray study of synthetic alunite. Acta Phys. Pol. A 1994, 86, 621–627. Available online: https://www.infona.pl/resource/bwmeta1.element.bwnjournal-article-appv86z420kz. [CrossRef]
- Luo, Z.Q.; Zhou, X.T.; Jia, Q.M.; Chen, X.F.; Tao, Z.C.; Liu, S.Q. Preparation of arsenical-natroalunite solid solutions with high crystallinity by hydrothermal method. Mater. Res. Innov. 2015, 19, 26–29. [Google Scholar] [CrossRef]
- Wang, L.; Xue, N.; Zhang, Y.; Hu, P. Controlled Hydrothermal Precipitation of Alunite and Natroalunite in High-Aluminum Vanadium-Bearing Aqueous System. Minerals 2021, 11, 892. [Google Scholar] [CrossRef]
- Zhu, Y.; Xuan, H.; Liang, Y.; Yan, Q.; Zhu, Z.; Jiang, Z.; Zhang, L.; Liu, L.; Tang, S. Dissolution and Solubility of the Synthetic Natroalunite and the Arsenic-Incorporated Natroalunite at pH of 2.00–5.60 and 25–45 °C. J. Chem. 2019, 2019, 9568360. [Google Scholar] [CrossRef]
- Cloutis, E.A.; Hawthorne, F.C.; Mertzman, S.A.; Krenn, K.; Craig, M.A.; Marcino, D.; Methot, M.; Strong, J.; Mustard, J.F.; Blaney, D.L.; et al. Detection and discrimination of sulfate minerals using reflectance spectroscopy. Icarus 2006, 184, 121–157. [Google Scholar] [CrossRef]
- Aguilar-Carrillo, J.; Villalobos, M.; Pi-Puig, T.; Escobar-Quiroz, I.N.; Romero, F.M. Synergistic arsenic (V) and lead (II) retention on synthetic jarosite. Simultaneous structural incorporation behaviour and mechanism. Enviromental. Sci. Process Impact 2018, 20, 354–369. [Google Scholar] [CrossRef]
- Szymanski, J.T. The Crystal structure of beudantite, Pb(Fe,Al)3[(As,S)O4](OH)6. Can. Mineral. 1988, 26, 923–932. [Google Scholar]
- Frost, R.L.; Wills, R.A.; Weier, M.L.; Martens, W.; Mills, S. A Raman spectroscopic study of selected natural jarosites. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 63, 18. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, Y.; Zhang, J.; Wang, Y.; Hickmott, D.D.; Daemen, L.L.; Hartl, M.A.; Wang, L. Anisotropic elasticity of jarosite: A high-P synchrotron XRD study. Am. Mineral. 2010, 95, 19–23. [Google Scholar] [CrossRef]
- Sunyer, A.; Viñals, J. Arsenate substitution in natroalunite: A Potential Mèdium for Arsenic Immobilization. Part 2: Cell Parameters and Stability Tests. Hydrometallurgy 2011, 109, 106–115. [Google Scholar] [CrossRef]
- Frost, R.L.; Wain, D. A Thermogravimetric and Infrared Emission Spectros-copic Study of Alunite. J. Therm. Anal. Calorim. 2008, 91, 267–274. [Google Scholar] [CrossRef]
- Drouet, C.; Navrotsky, A. Synthesis, characterization and thermochemistry of K-Na_H3O jarosites. Geochim. Cosmochim. Acta 2003, 67, 2063–2076. [Google Scholar] [CrossRef]
- Perez-Labra, M.; Romero-Serrano, A.; Salinas-Rodriguez, E.; Avila-Dávila, E.O.; Reyes-Pérez, M. Synthesis, thermodynamics and kinetics of rubidium jarosite decomposition in calcium hydroxide solutions. Metall. Mater. Trans. B 2013, 43, 773–780. [Google Scholar] [CrossRef]
- Forray, F.L.; Smith, A.M.L.; Drouet, C.; Navrotsky, A.; Wright, K.; Hudson-Edwards, K.A.; Dubbin, W.E. Synthesis, characterization and thermo-chemistry of a Pb-jarosite. Geochim. Cosmochim. Acta 2010, 74, 215–224. [Google Scholar] [CrossRef]
- Forray, F.L.; Smith, A.M.L.; Navrotsky, A.; Wright, K.; Hudson-Edwards, K.A.; Dubbin, W.E. Synthesis, Characterization and Thermochemistry of Synthetic Pb-As, Pb-Cu and Pb-Zn Jarosites. Geochim. Cosmochim. Acta 2014, 127, 107–119. [Google Scholar] [CrossRef]
- Patiño, F.; Viñals, J.; Roca, A.; Núñez, C. Alkaline decomposition-cyanidation kinetics of argentian plumbojarosite. Hydrometallurgy 1994, 34, 279–291. [Google Scholar] [CrossRef]
- Roca, A.; Cruells, M.; Patiño, F.; Rivera, I.; Plata, M. Kinetic model for the cyanidation of silver ammonium jarosite in NaOH medium. Hydrometallurgy 2006, 81, 15–23. [Google Scholar] [CrossRef]
- Patiño, F.; Cruells, M.; Roca, A.; Salinas, E.; Pérez, M. Kinetics of alkaline decomposition and cyanidation of argentian ammonium jarosite in lime medium. Hydrometallurgy 2003, 70, 153–161. [Google Scholar] [CrossRef]
- Salinas, E.; Roca, A.; Cruells, M.; Patiño, F.; Córdoba, D.A. Characterization and alkaline decomposition-cyanidation kinetics of industrial ammonium jarosite in NaOH media. Hydrometallurgy 2001, 60, 237–246. [Google Scholar] [CrossRef]
- Ordóñez, S.; Flores, M.U.; Patiño, F.; Reyes, I.A.; Islas, H.; Reyes, M.; Méndez, E.; Palacios, E.G. Kinetic Analysis of the Decomposition Reaction of the Mercury Jarosite in NaOH Medium. Int. J. Chem. Kinet. 2017, 49, 798–809. [Google Scholar] [CrossRef]
- Salinas, E.; Cerecedo, E.; Ramirez, M.; Patiño, F.; Pérez, M. Kinetics of Alkaline Decomposition and Cyaniding of Argentian Rubidium Jarosite in NaOH Medium. Metall. Mater. Trans. B-Process Metall. Mater. Proccess. Sci. 2012, 43, 1027–1033. [Google Scholar] [CrossRef]
- Cerecedo, E.; Salinas, E.; Longoria, L.C.; Carrillo, F.R.; Hernández, J. Kinetics Study of Alkaline Decomposition of Rubidium Jarosite in Ca(OH)2. In Proceedings of the 2011 TMS Annual Meeting & Exhibition, Symposium: Hydrometallurgy Fundamentals and Applications, San Diego, CA, USA, 27 February–3 March 2011. [Google Scholar] [CrossRef]
- Reyes, I.A.; Mireles, I.; Patiño, F.; Pandiyan, T.; Flores, M.U.; Palacios, E.G.; Gutiérrez, E.J.; Reyes, M. A study on the dissolution rates of K-Cr(VI)-jarosites: Kinetic Analysis and Implications. Geochem. Trans. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Patiño, F.; Reyes, I.A.; Flores, M.U.; Pandiyan, T.; Roca, A.; Reyes, M.; Hernández, J. Kinetic modeling and experimental design of the sodium arsenojarosite decomposition in alkaline media: Implications. Hydrometallurgy 2013, 137, 115–125. [Google Scholar] [CrossRef]
- Patiño, F.; Flores, M.U.; Reyes, I.A.; Reyes, M.; Hernández, J.; Rivera, I.; Juárez, J.C. Alkaline decomposition of synthetic jarosite with arsenic. Geochem. Trans. 2013, 14, 2. Available online: https://geochemicaltransactions.biomedcentral.com/articles/10.1186/1467-4866-14-2. (accessed on 24 March 2022). [CrossRef] [PubMed]
- Patiño, F.; Flores, M.U.; Reyes, I.A.; Ordóñez, S.; Méndez, J.E.; Flores, V.H.; Islas, H.; Reyes, M. Kinetic modeling of the decomposition of beudantite in NaOH medium. React. Kinet. Mech. Catal. 2016, 119, 367–379. [Google Scholar] [CrossRef]
- Viñals, J.; Roca, A.; Cruells, M.; Núñez, C. Recovery of Gold and Silver from Plumbojarosite-Containing Hematite Tailings by Alkaline Pretreatment and Cyanidation. In EMC’91: Non-Ferrous Metallurgy, Present and Future; Springer: Dordrecht, The Netherlands, 1991; pp. 11–18. [Google Scholar]
- González-Ibarra, A.A.; Nava-Alonso, F.; Fuentes-Aceituno, J.C.; Uribe-Salas, A. Hydrothermal decomposition of industrial jarosite in alkaline media: The Rate Determining Step of the Process Kinetics. J. Min. Metall. Sect. B Metall. 2016, 52, 135–142. [Google Scholar] [CrossRef]
- Celep, O.; Serbest, V. Characterization of an iron oxy/hydroxide (gossan type) bearing refractory gold and silver ore by diagnostic leaching. Trans. Nonferrous Met. Soc. China 2015, 25, 1286–1297. [Google Scholar] [CrossRef]
- Viñals, J.; Núñez, C. Dissolution kinetics of argentian plumbojarosite from old tailings of sulfatizing roasting pyrites by HCl-CaCl2 leaching. Metall. Trans. B 1988, 19, 365–373. [Google Scholar] [CrossRef]
- Kendall, M.R.; Madden, A.S.; Madden, M.E.; Hu, Q. Effects of arsenic incorporation on jarosite dissolution rates and reaction products. Geochim. Cosmochim. Acta 2013, 112, 192–207. [Google Scholar] [CrossRef]
- Islas, H.; Flores, M.U.; Reyes, I.A.; Juárez, J.C.; Reyes, M.; Teja, A.M.; Palacios, E.G.; Pandiyan, T.; Aguilar-Carrillo, J. Determination of the dissolution rate of hazardous jarosites in different conditions using the shrinking core kinetic model. J. Hazard. Mater. 2020, 386, 121664. [Google Scholar] [CrossRef] [PubMed]
- Calla-Choque, D.; Lapidus, G.T. Jarosite dissolution kinetics in the presence of acidic thiourea and oxalate media. Hydrometallurgy 2021, 200, 105565. [Google Scholar] [CrossRef]
- Castro, L.; Blázquez, M.L.; González, F.; Muñoz, J.A.; Ballester, A. Anaerobic bioleaching of jarosites by Shewanella putrefaciens, influence of chelators and biofilm formation. Hydrometallurgy 2017, 168, 56–63. [Google Scholar] [CrossRef]
- Castro, L.; Blázquez, M.L.; González, F.; Muñoz, J.A.; Ballester, A. Anaerobic Bioreduction of Jarosites and Biofilm Formation by a Natural Microbial Consortium. Minerals 2019, 9, 81. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Sunyer, A. Hematite formation from jarositetype compounds by hidrotermal conversion. Can. Metall. Q. 2012, 51, 11–23. [Google Scholar] [CrossRef]
- Frost, R.L.; Wills, R.A.; Weier, M.L.; Musumeci, A.W.; Martens, W. Thermal decomposition of natural and synthetic plumbojarosite: Importance in “Archeochemistry”. Thermochim. Acta 2005, 432, 30–35. [Google Scholar] [CrossRef]
- Frost, R.L.; Wills, R.A.; Kloprogge, J.T.; Martens, W. Thermal decomposition of hydronium jarosite (H3O)Fe3(SO4)2(OH)6. J. Therm. Anal. Calorim. 2006, 83, 213–218. [Google Scholar] [CrossRef]
- Vu, H.N.; Dvořák, P.; Síta, T. Study of conversion of waste jarosite precipitates to hematite. Inz. Miner. 2014, 15, 275–280. Available online: http://www.potopk.com.pl/Full_text/2014_full/2014_2_46.pdf.
- Ma, X.; Tan, H.; Dong, F.; Li, B.; Liu, J.; Chen, Y.; Wang, L. Preparation of Pyrrhotite from Ammonium Jarosite and Estimation of Activation Energy in Reducing Atmosphere. Int. J. Chem. React. Eng. 2019, 17, 20180149. [Google Scholar] [CrossRef]
- Ma, X.; Tan, H.; Dong, F.; Li, B.; Wang, J.; He, X.; Liu, C. Preparation of ammonium jarosite and estimated activation energy of thermal decomposition in reducing atmosphere. J. Therm. Anal. Calorim. 2019, 135, 2565–2572. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, H.; Hou, X.; Gao, W.; Gui, H.; Liu, Q. The effect of pellet technology on direct reduction of jarosite residues from zinc hydrometallurgy. Physicochem. Probl. Miner. Proccess. 2019, 55, 802–811. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, H.; Zhang, G.; Kang, J.; Wang, C. Comprehensive recovery and recycle of jarosite residues from zinc hydrometallurgy. Chem. Eng. J. Adv. 2020, 3, 100023. [Google Scholar] [CrossRef]
- Kolitsch, U.; Pring, A. Crystal chemistry of the crandallite, beudantite and alunite groups: A Review and Evaluation of the Suitability as Storage Materials for Toxic Metals. J. Mineral. Petrol. Sci. 2001, 96, 67–78. [Google Scholar] [CrossRef]
- Roussel, C.; Neel, C.; Bril, H. Minerals controlling arsenic and lead solubility in an abandoned gold mine tailings. Sci. Total Environ. 2000, 263, 209–219. [Google Scholar] [CrossRef]
- Smeaton, C.M.; Walshe, G.E.; Smith, A.M.L.; Hudson-Edwards, K.A.; Dubbin, W.E.; Wright, K.; Beale, A.M.; Fryer, B.J.; Weisener, C.G. Simultaneous Release of Fe and as during the Reductive Dissolution of Pb−As Jarosite by Shewanella putrefaciens CN32. Environ. Sci. Technol. 2012, 46, 12823–12831. [Google Scholar] [CrossRef]
- Alarcón, R.; Gaviria, J.; Dold, B. Liberation of Adsorbed and Co-Precipitated Arsenic from Jarosite, Schwertmannite, Ferrihydrite, and Goethite in Seawater. Minerals 2014, 4, 603–620. [Google Scholar] [CrossRef]
- Zhu, Y.; Wei, W.; Tang, S.; Zhu, Z.; Yan, Q.; Zhang, L.; Deng, H. A comparative study on the dissolution and stability of beudantite and hidalgoite at pH 2–12 and 25–45 °C for the possible long-term simultaneous immobilization of arsenic and lead. Chemosphere 2021, 263, 128386. [Google Scholar] [CrossRef]
- Hudson-Edwards, K.A. Uptake and release of arsenic and antimony in alunite–jarosite and beudantite group minerals. Am. Mineral. 2019, 104, 5. [Google Scholar] [CrossRef]
- Johnston, S.G.; Burton, E.D.; Keene, A.F.; Planer-Friedrich, B.; Voegelin, A.; Blackford, M.G.; Lumpkin, G.R. Arsenic mobilization and iron transformations during sulfidization of As(V)-bearing jarosite. Chem. Geol. 2012, 334, 9–24. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Passivation of chalcopyrite durin gits chemical leaching with ferric ion at 68 °C. Miner. Eng. 2009, 22, 229–235. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Comparative kinetic study of the silver-catalyzed chalcopyrite leaching at 35 °C and 68 °C. Int. J. Miner. Process. 2009, 92, 137–143. [Google Scholar] [CrossRef]
- Kartal, M.; Xia, F.; Ralph, D.; Rickard, W.D.A.; Renard, F.; Li, W. Enhancing chalcopyrite leaching by tetrachloroethylene-assisted removal of sulfur passivation and the mechanisms of jarosite formation. Hydrometallurgy 2020, 191, 105192. [Google Scholar] [CrossRef]
- Quezada, V.; Roca, A.; Benavente, O.; Cruells, M.; Melo, E. The effects of sulphuric acid and sodium chloride agglomeration and curing on chalcopyrite leaching. Metals 2021, 11, 873. [Google Scholar] [CrossRef]
- Quezada, V.; Roca, A.; Benavente, O.; Cruells, M.; Melo, E.; Hernández, M. Pretreatment to leaching for a primary copper sulphide ore in chloride media. Metals 2021, 11, 1260. [Google Scholar] [CrossRef]
- Sasaki, K.; Nakamuta, Y.; Hirajima, T.; Tuovinen, O.H. Raman characterization of secondary minerals formed during chalcopyrite leaching with Acidithiobacillus ferrooxidans. Hydrometallurgy 2009, 95, 153–158. [Google Scholar] [CrossRef]
- Vakylabad, A.B.; Nazari, S.; Darezereshki, E. Bioleaching of copper from chalcopyrite ore at higher NaCl concentrations. Miner. Eng. 2022, 175, 107281. [Google Scholar] [CrossRef]
- Morris, R.V.; Golden, D.C.; Bell III, J.F.; Shelfer, T.D.; Scheinost, A.C.; Hinman, N.W.; Furniss, G.; Mertzman, S.A.; Bishop, J.L.; Ming, D.W.; et al. Mineralogy, composition, and alteration of Mars Pathfinder Rocks and soils’ Evidence from multispectral, elemental, and magnetic data on terrestrial analogue, SNC meteorite, and Pathfinder samples. J. Geophys. Res. 2000, 105, 1757–1817. [Google Scholar] [CrossRef]
- Klingelhöfer, G.; Morris, R.V.; Bernhardt, B.; Schröder, C.; Rodionov, D.S.; de Souza, P.A.; Yen, A.; Gellert, R.; Evlanov, E.N.; Zubkov, B.; et al. Jarosite and Hematite at Meridiani Planum from Opportunity’s Mössbauer Spectrometer. Science 2004, 306, 1740–1745. [Google Scholar] [CrossRef]
- Madden, M.E.E.; Madden, A.S.; Rimstidt, J.D.; Zahrai, S.; Kendall, M.R.; Miller, M.A. Jarosite dissolution rates and nanoscale mineralogy. Geochim. Cosmochim. Acta 2013, 91, 306–321. [Google Scholar] [CrossRef]
- Rampe, E.B.; Ming, D.W.; Blake, D.F.; Bristow, T.F.; Chipera, S.J.; Grotzinger, J.P.; Morris, R.V.; Morrison, S.M.; Vaniman, D.T.; Yen, A.S.; et al. Mineralogy of an ancient lacustrine mudstone succession from the Murray formation, Gale crater, Mars. Earth Planet. Sci. Lett. 2017, 471, 172–185. [Google Scholar] [CrossRef]
- Potter-McIntyre, S.L.; McCollom, T.M. Jarosite and Alunite in Ancient Terrestrial Sedimentary Rocks: Reinterpreting Martian Depositional and Diagenetic Environmental Conditions. Life 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, L.; Fernández-Martínez, M.A.; Moreno-Paz, M.; Carrizo, D.; García-Villadangos, M.; Manchado, J.M.; Stoker, C.R.; Glass, B.; Parro, V. Simulating Mars Drilling Mission for Searching for Life: Ground-Truthing Lipids and Other Complex Microbial Biomarkers in the Iron-Sulfur Rich Río Tinto Analog. Astrobiology 2020, 20, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Baccolo, G.; Delmonte, B.; Niles, P.B.; Cibin, G.; Di Stefano, E.; Hampai, D.; Keller, L.; Maggi, V.; Marcelli, A.; Michalski, J.; et al. Jarosite formation in deep Antarctic ice provides a window into acidic, water-limited weathering on Mars. Nat. Commun. 2021, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Grasby, S.E.; Percival, J.B.; Bilot, I.; Ardakani, O.H.; Smith, I.R.; Galloway, J.; Bringué, M.; McLoughlin-Coleman, T. Extensive jarosite deposits formed through auto-combustion and weathering of pyritiferous mudstone, Smoking Hills (Ingniryuat), Northwest Territories, Canadian Arctic—A potential Mars analogue. Chem. Geol. 2022, 587, 2022. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).