

The Role of the Metal in the Catalytic Reactions of Hydrogenation–Dehydrogenation of Polycyclic Hydrocarbons for Hydrogen Storage


Abstract
:1. Introduction
2. Catalytic Hydrogenation and Dehydrogenation of Cyclic Hydrocarbons on Supported Metals
2.1. Benzene–Cyclohexane
2.2. Toluene–Methylcyclohexane
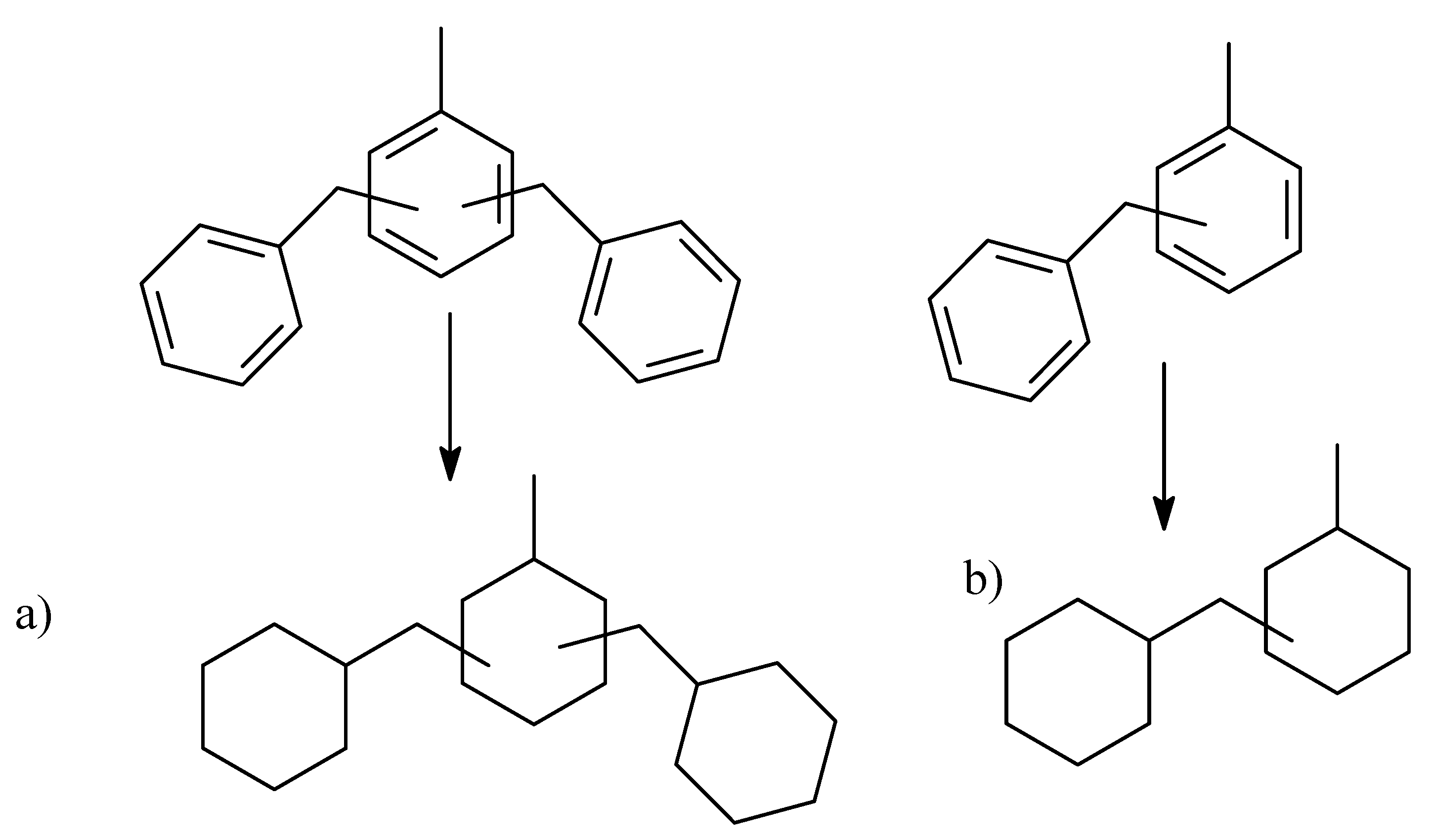
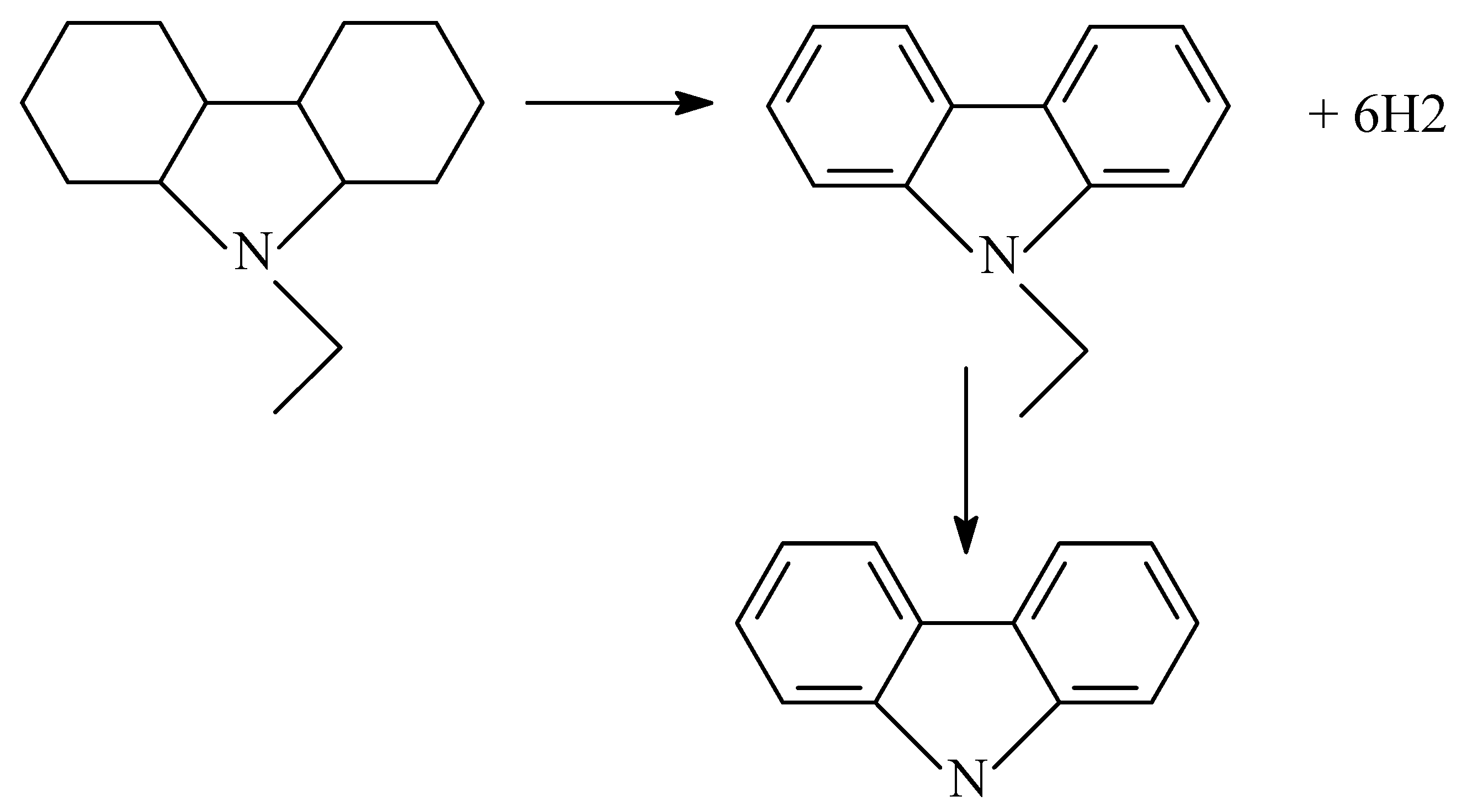
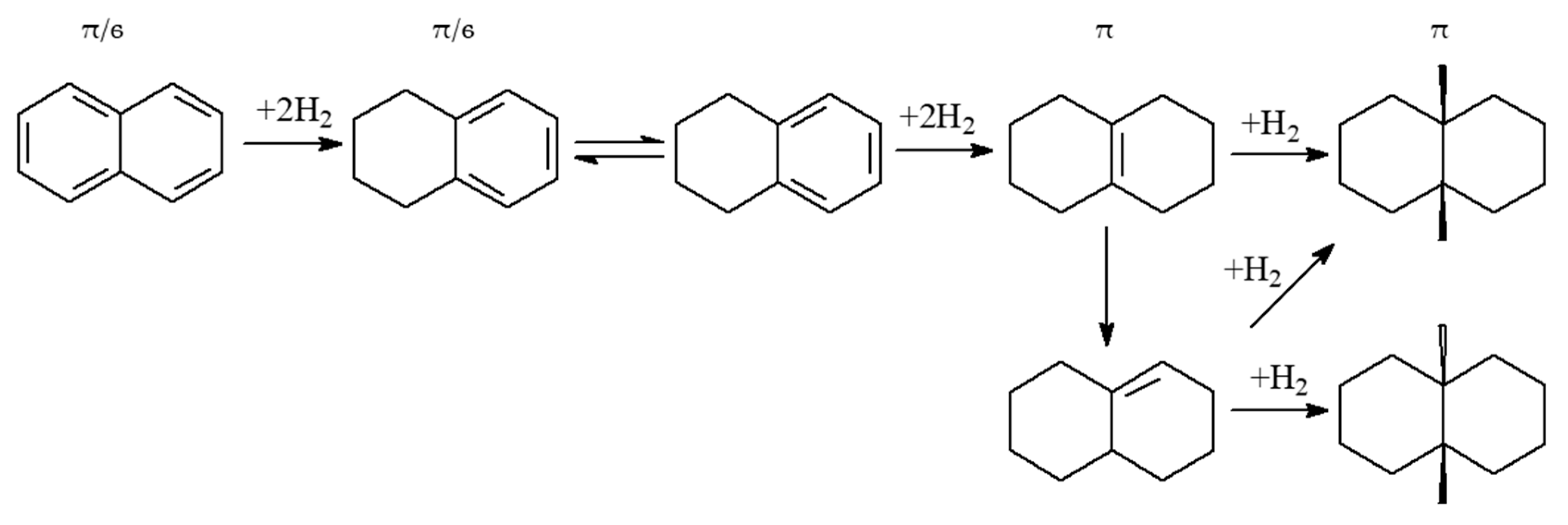
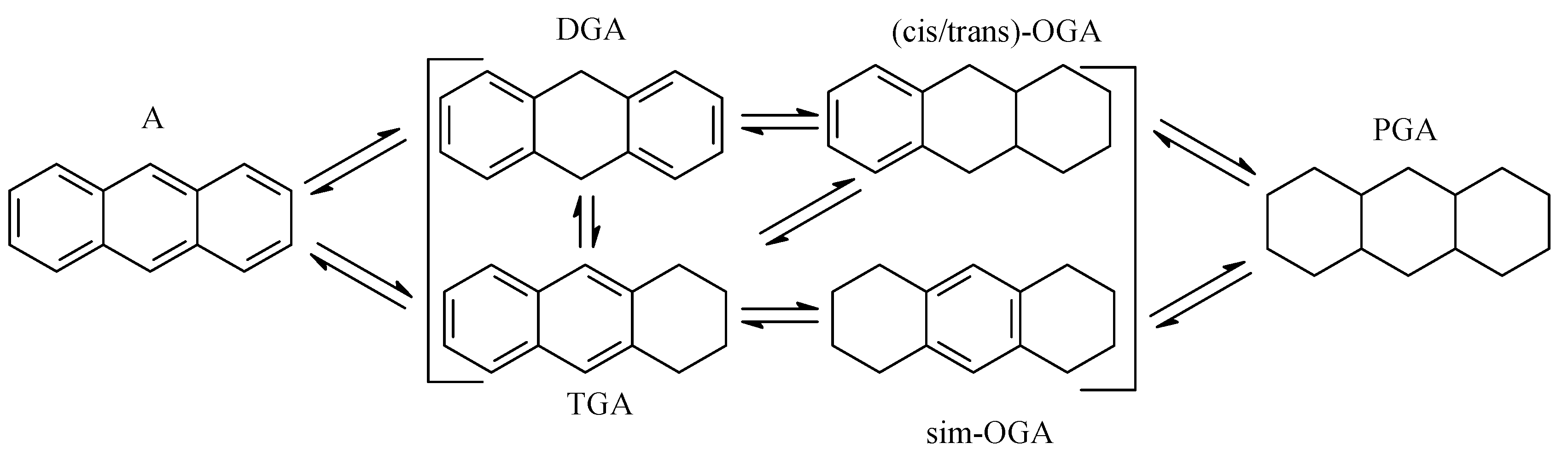
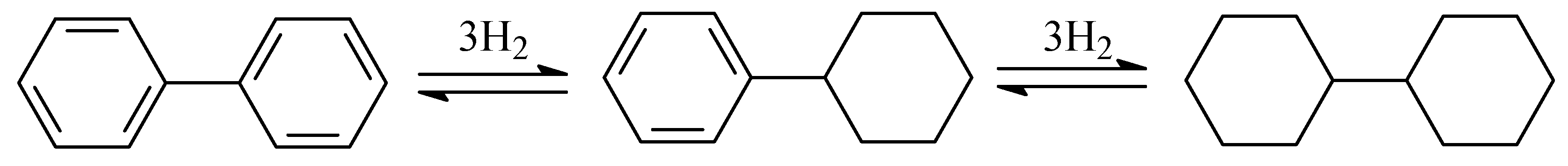
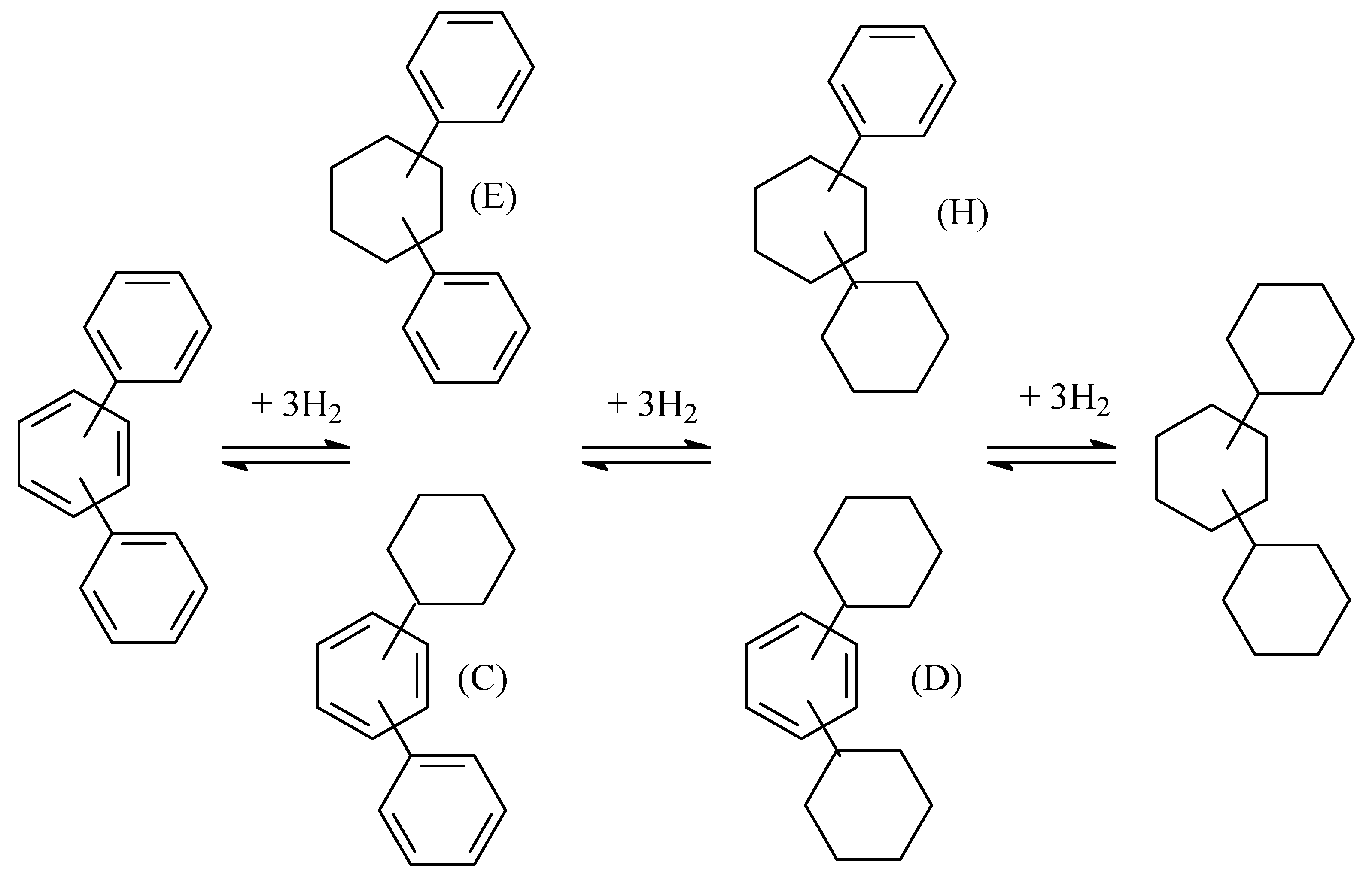
2.3. Polycyclic Hydrocarbons
3. Conclusions and Outlooks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Balali, Y.; Stegen, S. Review of energy storage systems for vehicles based on technology, environmental impacts, and costs. Renew. Sustain. Energy Rev. 2021, 135, 110185. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, N.; Hillmansen, S.; Roberts, C.; Yan, Y. Techno-Economic Analysis of Hydrogen Storage Technologies for Railway Engineering: A Review. Energies 2022, 15, 6467. [Google Scholar] [CrossRef]
- Li, M.; Bai, Y.; Zhang, C.; Song, Y.; Jiang, S.; Grouset, D.; Zhang, M. Review on the research of hydrogen storage system fast refueling in fuel cell vehicle. Int. J. Hydrog. Energy 2019, 44, 10677–10693. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Musyoka, N.M.; Langmi, H.W.; Mathe, M.; Liao, S. Current research trends and perspectives on materials-based hydrogen storage solutions: A critical review. Int. J. Hydrog. Energy 2017, 42, 289–311. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Rayalu, S.; Devotta, S.; Ichikawa, M. Chemical hydrides: A solution to high capacity hydrogen storage and supply. Int. J. Hydrog. Energy 2008, 33, 360–365. [Google Scholar] [CrossRef]
- Bourane, A.; Elanany, M.; Pham, T.V.; Katikaneni, S.P. An overview of organic liquid phase hydrogen carriers. Int. J. Hydrog. Energy 2016, 41, 23075–23091. [Google Scholar] [CrossRef]
- Singla, M.K.; Nijhawan, P.; Oberoi, A.S. Hydrogen fuel and fuel cell technology for cleaner future: A review. Environ. Sci. Pollut. Res. 2021, 28, 15607–15626. [Google Scholar] [CrossRef]
- von Helmolt, R.; Eberle, U. Fuel cell vehicles: Status 2007. J. Power Sources 2007, 165, 833–843. [Google Scholar] [CrossRef]
- Makaryan, I.A.; Sedov, I.V.; Maksimov, A.L. Hydrogen Storage Using Liquid Organic Carriers. Russ. J. Appl. Chem. 2020, 93, 1815–1830. [Google Scholar] [CrossRef]
- Jorschick, H.; Geißelbrecht, M.; Eßl, M.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Benzyltoluene/dibenzyltoluene-based mixtures as suitable liquid organic hydrogen carrier systems for low temperature applications. Int. J. Hydrog. Energy 2020, 45, 14897–14906. [Google Scholar] [CrossRef]
- Stroman, R.O.; Schuette, M.W.; Swider-Lyons, K.; Rodgers, J.A.; Edwards, D.J. Liquid hydrogen fuel system design and demonstration in a small long endurance air vehicle. Int. J. Hydrog. Energy 2014, 39, 11279–11290. [Google Scholar] [CrossRef]
- Geburtig, D.; Preuster, P.; Bösmann, A.; Müller, K.; Wasserscheid, P. Chemical utilization of hydrogen from fluctuating energy sources—Catalytic transfer hydrogenation from charged Liquid Organic Hydrogen Carrier systems. Int. J. Hydrog. Energy 2016, 41, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Kustov, L.M.; Kalenchuk, A.N.; Bogdan, V.I. Systems for accumulation, storage and release of hydrogen. Russ. Chem. Rev. 2020, 89, 897–916. [Google Scholar] [CrossRef]
- Sekine, Y.; Higo, T. Recent Trends on the Dehydrogenation Catalysis of Liquid Organic Hydrogen Carrier (LOHC): A Review. Top. Catal. 2021, 64, 470–480. [Google Scholar] [CrossRef]
- Cho, J.-Y.; Kim, H.; Oh, J.-E.; Park, B.Y. Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems. Catalysts 2021, 11, 1497. [Google Scholar] [CrossRef]
- Rao, P.C.; Yoon, M. Potential Liquid-Organic Hydrogen Carrier (LOHC) Systems: A Review on Recent Progress. Energies 2020, 13, 6040. [Google Scholar] [CrossRef]
- Jorschick, H.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Hydrogenation of aromatic and heteroaromatic compounds—A key process for future logistics of green hydrogen using liquid organic hydrogen carrier systems. Sustain. Energy Fuels 2021, 5, 1311–1346. [Google Scholar] [CrossRef]
- Bulgarin, A.; Jorschick, H.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Purity of hydrogen released from the Liquid Organic Hydrogen Carrier compound perhydro dibenzyltoluene by catalytic dehydrogenation. Int. J. Hydrog. Energy 2019, 45, 712–720. [Google Scholar] [CrossRef]
- Pez, G.P.; Scott, A.R.; Cooper, A.C.; Cheng, H. Hydrogen storage by reversible hydrogenation of π-conjugated substrates. US Patent 7,101,530, 2005. Air Products and Chemicals, Inc.: Allentown, PA, USA. [Google Scholar]
- Ribeiro da Silva, M.A.V.; Santos, L.M.N.B.F.; Spencer, S.; Lima, L.M. Standard molar enthalpies of formation and of sublimation of the terphenyl isomers. J. Chem. Thermodyn. 2008, 40, 375–384. [Google Scholar] [CrossRef]
- Allison, T.C.; Donald, R.; Burgess, D.R., Jr. First-Principles Prediction of Enthalpies of Formation for Polycyclic Aromatic Hydrocarbons and Derivatives. J. Phys. Chem. A 2015, 119, 11329–11365. [Google Scholar] [CrossRef]
- Alberty, R.A.; Reif, A.K. Standard Chemical Thermodynamic Properties of Polycyclic Aromatic Hydrocarbons and Their Isomer Groups I. Benzene Series. J. Phys. Chem. Ref. Data 1988, 17, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Saeys, M.; Reyniers, M.F.; Neurock, M.; Marin, G.B. An Initio Reaction Path Analayis of Benzene Hydrogenation to Cyclohexane on Pt(111). J. Phys. Chem. B 2005, 109, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Saeys, M.; Reyniers, M.-F.; Thybaut, J.W.; Neurock, M.; Marin, G.B. First-principles based kinetic model for the hydrogenation of toluene. J. Catal. 2005, 236, 129–138. [Google Scholar] [CrossRef]
- Balandin, A.A. The Multiplet Theory of Catalysis. Structural Factors in Catalysis. Russ. Chem. Rev. 1962, 31, 589–614. [Google Scholar] [CrossRef]
- Campbell, J.M.; Seimanides, S.; Campbell, C.T. ChemInform Abstract: Probing Ensemble Effects in Surface Reactions. Part 2. Benzene Adsorption on Clean and Bismuth-Covered Pt(111). J. Phys. Chem. 1989, 20, 815–826. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Campbell, C.T. ChemInform Abstract: Probing Ensemble Effects in Surface Reactions. Part 3. Cyclohexane Adsorption on Clean and Bismuth-Covered Pt(111). J. Phys. Chem. 1989, 20, 826–835. [Google Scholar] [CrossRef]
- Henn, F.C.; Dalton, P.J.; Campbell, C.T. ChemInform Abstract: Probing Ensemble Effects in Surface Reactions. Part 4. Cyclopentene Adsorption on Clean and Bismuth-Covered Pt(111). J. Phys. Chem. 1989, 20, 836–846. [Google Scholar] [CrossRef]
- Kariya, N.; Fukuoka, A.; Utagawa, T.; Sakuramoto, M.; Goto, Y.; Ichikawa, M. Efficient hydrogen production using cyclohexane and decalin by pulse-spray mode reactor with Pt catalysts. Appl. Catal. A Gen. 2003, 247, 247–259. [Google Scholar] [CrossRef]
- Kariya, N.; Fukuoka, A.; Ichikawa, M. Efficient evolution of hydrogen from liquid cycloalkanes over Pt-containing catalysts supported on active carbons under “wet–dry multiphase conditions”. Appl. Catal. A Gen. 2002, 233, 91–102. [Google Scholar] [CrossRef]
- Smith, C.E.; Biberian, J.P.; Somorjai, G.A. The Effect of Strogly Bound Oxygen on the Dehydrogenation and Hydrogenation Activity and Selectivity of Platinum Single Crystal Surfaces. J. Catal. 1979, 57, 426–443. [Google Scholar] [CrossRef]
- Cromwell, D.; Vasudevan, P.; Pawelec, B.; Fierro, J.L.G. Enhanced methylcyclohexane dehydrogenation to toluene over Ir/USY catalyst. Catal. Today 2016, 259, 119–129. [Google Scholar] [CrossRef]
- Takise, K.; Sato, A.; Murakami, K.; Ogo, S.; Gil Seo, J.; Imagawa, K.-I.; Kado, S.; Sekine, Y. Irreversible catalytic methylcyclohexane dehydrogenation by surface protonics at low temperature. RSC Adv. 2019, 9, 5918–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, M.; Cresswell, D.; Garforth, A. Detailed reaction kinetics for the dehydrogenation of methylcyclohexane over Pt catalyst. Ind. Eng. Chem. Res. 2012, 51, 158–170. [Google Scholar] [CrossRef]
- Van Trimpont, P.A.; Marin, G.B.; Froment, G.F. Activities and Selectivities for Reforming Reactions on Un-Sulfided and Sulfided Commercial Platinum and Platinum-Rhenium Catalysts. Appl. Catal. A Gen. 1985, 17, 161–173. [Google Scholar] [CrossRef]
- Johnson, A.L.; Muetterties, E.L.; Stoehr, J. ChemInform Abstract: Orientation of Complex Molecules Chemisorbed on Metal Surfaces: Near-Edge X-Ray Absorption Studies. J. Am. Chem. Soc. 1983, 105, 7183–7185. [Google Scholar] [CrossRef]
- Steinrück, H.-P.; Fuhrmann, T.; Papp, C.; Tränkenschuh, B.; Denecke, R. A detailed analysis of vibrational excitations in x-ray photoelectron spectra of adsorbed small hydrocarbons. J. Chem. Phys. 2006, 125, 204706. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Nobuko, K.; Masaru, I. Dehydrogenation of cyclohexane over Ni based catalysts supported on activated carbon using spray pulsed reactor and enhancement in activity by addition of a small amount of Pt. Catal. Lett. 2005, 105, 83–87. [Google Scholar] [CrossRef]
- Okada, Y.; Sasaki, E.; Watanabe, E.; Hyodo, S.; Nishijima, H. Development of dehydrogenation catalyst for hydrogen generation in organic chemical hydride method. Int. J. Hydrog. Energy 2006, 31, 1348–1356. [Google Scholar] [CrossRef]
- Okada, Y.; Imagawa, K.; Shimura, M. Hydrogen storage and transportation system in large-scale—SPERA hydrogen system. Fuel Cell Technol. 2014, 14, 36–40. [Google Scholar]
- Amende, M.; Gleichweit, C.; Xu, T.; Höfert, O.; Koch, M.; Wasserscheid, P.; Steinrück, H.-P.; Papp, C.; Libuda, J. Dicyclohexylmethane as a Liquid Organic Hydrogen Carrier: A Model Study on the Dehydrogenation Mechanism over Pd(111). Catal. Lett. 2016, 146, 851–860. [Google Scholar] [CrossRef]
- Saito, Y. Powerful H2 supply from organic hydride and its circulation system for hydrogen transportation. Catal. Catal. 2005, 47, 137–139. [Google Scholar]
- Sotoodeh, F.; Zhao, L.; Smith, K.J. Kinetics of H2 recovery from dodecahydro-N-ethylcarbazole over a supported Pd catalyst. Appl. Catal. A Gen. 2009, 362, 155–162. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Smith, K.J. Kinetics of Hydrogen Uptake and Release from Heteroaromatic Compounds for Hydrogen Storage. Ind. Eng. Chem. Res. 2010, 49, 1018–1026. [Google Scholar] [CrossRef]
- Crawford, P.; Burch, R.; Hardacre, C.; Hindle, K.T.; Hu, P.; Kalirai, A.B.; Rooney, D.W. Understanding the Dehydrogenation Mechanism of Tetrahydrocarbazole over Palladium Using a Combined Experimental and Density Functional Theory Approach. J. Phys. Chem. C 2007, 111, 6434–6439. [Google Scholar] [CrossRef]
- Hindle, K.; Burch, R.; Crawford, P.; Hardacre, C.; Hu, P.; Kalirai, B.; Rooney, D. Dramatic liquid-phase dehydrogenation rate enhancements using gas-phase hydrogen acceptors. J. Catal. 2007, 251, 338–344. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Huber, B.J.M.; Smith, K.J. Dehydrogenation kinetics and catalysis of organic heteroaromatics for hydrogen storage. Int. J. Hydrog. Energy 2012, 37, 2715–2722. [Google Scholar] [CrossRef]
- Gleichweit, C.; Amende, M.; Schernich, S.; Zhao, W.; Lorenz, M.P.A.; Höfert, O.; Brückner, N.; Wasserscheid, P.; Libuda, J.; Steinrück, H.-P.; et al. Dehydrogenation of Dodecahydro-N-ethylcarbazole on Pt(111). ChemSusChem 2013, 6, 974–977. [Google Scholar] [CrossRef]
- Amende, M.; Gleichweit, C.; Werner, K.; Schernich, S.; Zhao, W.; Lorenz, M.P.A.; Höfert, O.; Papp, C.; Koch, M.; Wasserscheid, P.; et al. Model Catalytic Studies of Liquid Organic Hydrogen Carriers: Dehydrogenation and Decomposition Mechanisms of Dodecahydro-N-ethylcarbazole on Pt(111). ACS Catal. 2014, 4, 657–665. [Google Scholar] [CrossRef]
- Amende, M.; Gleichweit, C.; Schernich, S.; Höfert, O.; Lorenz, M.P.A.; Zhao, W.; Koch, M.; Obesser, K.; Papp, C.; Wasserscheid, P.; et al. Size and Structure Effects Controlling the Stability of the Liquid Organic Hydrogen Carrier Dodecahydro-N-ethylcarbazole during Dehydrogenation over Pt Model Catalysts. J. Phys. Chem. Lett. 2014, 5, 1498–1504. [Google Scholar] [CrossRef]
- Rautanen, P.; Lylykangas, M.; Aittamaa, J.; Krause, A. Liquid Phase Hydrogenation of Naphthalene on Ni/Al2O3. Stud. Surf. Sci. Catal. 2001, 133, 309–316. [Google Scholar]
- Rautanen, P.A.; Lylykangas, M.S.; Aittamaa, J.R.; Krause, A.O.I. Liquid-Phase Hydrogenation of Naphthelene and Tetralin on Ni/Al2O3. Kinetic Modeling. Ind. Eng. Chem. Res. 2002, 41, 5966–5975. [Google Scholar] [CrossRef]
- Li, P.; Huang, Y.-L.; Chen, D.; Zhu, J.; Zhao, T.-J.; Zhou, X.-G. CNFs-supported Pt catalyst for hydrogen evolution from decalin. Catal. Commun. 2009, 10, 815–818. [Google Scholar] [CrossRef]
- Weitkamp, A. Deuteriation and deuterogenation of naphthalene and two octalins. J. Catal. 1966, 6, 431–457. [Google Scholar] [CrossRef]
- Weitkamp, A. Stereochemistry and Mechanism of Hydrogenation of Naphthalenes on Transition Metal Catalysts and Conformational Analysis of the Products. Adv. Catal. 1968, 18, 1–110. [Google Scholar]
- Resini, C.; Lucarelli, C.; Taillades-Jacquin, M.; Liew, K.-E.; Gabellini, I.; Albonetti, S.; Wails, D.; Rozie`re, J.; Vaccari, A.; Jones, D. Pt-Sn/γ-Al2O3 and Pt-Sn-Na/γ-Al2O3 catalysts for hydrogen production by dehydrogenation of Jet A-1 fuel: Characterisation and preliminary activity tests. Int. J. Hydrog. Energy 2011, 36, 5972–5982. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Froment, G.F.; Goodman, D.W. CO-free hydrogen production via dehydrogenation of a Jet A hydrocarbon mixture. J. Catal. 2008, 253, 239–243. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Smetneva, D.N.; Bogdan, V.I.; Kustov, L. Kinetics of decalin dehydrogenation on Pt/C catalyst. Rus. Chem. Bull. 2015, 64, 2642–2645. [Google Scholar] [CrossRef]
- Hodoshima, S.; Nagata, H.; Saito, Y. Efficient hydrogen supply from tetralin with superheated liquid-film-type catalysis for operating fuel cells. Appl. Catal. A Gen. 2005, 292, 90–96. [Google Scholar] [CrossRef]
- Hodoshima, S.; Takaiwa, S.; Shono, A.; Satoh, K.; Saito, Y. Hydrogen storage by decalin/naphthalene pair and hydrogen supply to fuel cells by use of superheated liquid-film-type catalysis. Appl. Catal. A Gen. 2005, 283, 235–242. [Google Scholar] [CrossRef]
- Lázaro, M.; García-Bordejé, E.; Sebastián, D.; Moliner, R. In situ hydrogen generation from cycloalkanes using a Pt/CNF catalyst. Catal. Today 2008, 138, 203–209. [Google Scholar] [CrossRef]
- Okada, Y.; Imagawa, K.; Kawai, H. Safety of H2 storage and transportation technology of H2 energy in large-scale. Petrotech 2015, 38, 660–664. [Google Scholar]
- Nanzhe, J.K.S.; Rao, R.; Jin, M.-J.; Park, S.-E. Effect of hydrogen spillover in decalin dehydrogenation over supported Pt catalysts. Appl. Catal. A Gen. 2012, 425–426, 62–67. [Google Scholar]
- Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis; John Willey & Sons, Inc.: Hoboken, NJ, USA, 2001; 737p. [Google Scholar]
- Sun, L.-B.; Wei, X.-Y.; Liu, X.-Q.; Zong, Z.-M.; Li, W.; Kou, J.-H. Selective Hydrogen Transfer to Anthracene and Its Derivatives over an Activated Carbon. Energy Fuels 2009, 23, 4877–4882. [Google Scholar] [CrossRef]
- Ma, Y.-M.; Wei, X.-Y.; Zhou, X.; Cai, K.-Y.; Peng, Y.-L.; Xie, R.-L.; Zong, Y.; Wei, Y.-B.; Zong, Z.-M. Microwave-Assisted Hydrogen Transfer to Anthracene and Phenanthrene over Pd/C. Energy Fuels 2009, 23, 638–645. [Google Scholar] [CrossRef]
- Pinilla, J.; García, A.; Philippot, K.; Lara, P.; García-Suárez, E.; Millan, M. Carbon-supported Pd nanoparticles as catalysts for anthracene hydrogenation. Fuel 2014, 116, 729–735. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Koklin, A.E.; Bogdan, V.I.; Lunin, V.V. Hydrogenation of Anthracene and Dehydrogenation of Perhydroanthracene on Pt/C Catalysts. Russ. J. Phys. Chem. A 2018, 92, 663–668. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Koklin, A.E.; Bogdan, V.I.; Kustov, L.M. Hydrogenation of naphthalene and anthracene on Pt/C catalysts. Rus. Chem. Bull. 2018, 67, 1406–1411. [Google Scholar] [CrossRef]
- Abu-Rezig, R.; Avnir, D.; Moloslavski, I.; Schumann, H.; Blum, J. Entrapment of metallic palladium and a rodium (I) complex in silica sol-gel matrix. Formation of a highly active recyclable arene hydrogenation catalyst. J. Mol. Catal. A Chem. 2002, 12, 179–185. [Google Scholar] [CrossRef]
- Park, I.S.; Kwon, M.S.; Kang, K.Y.; Lee, J.S.; Park, J. Rhodium and Iridium Nanoparticles Entrapped in Aluminum Oxyhydroxide Nanofibers: Catalysts for Hydrogenations of Arenes and Ketones at Room Temperature with Hydrogen Balloon. Adv. Synth. Catal. 2007, 349, 2039–2047. [Google Scholar] [CrossRef]
- Jacinto, M.J.; Santos, O.H.; Landers, R.; Kiyohara, P.K.; Rossi, L.M. On the catalytic hydrogenation of polycyclic aromatic hydrocarbons into less toxic compounds by a facile recoverable catalyst. Appl. Catal. B Environ. 2009, 90, 688–692. [Google Scholar] [CrossRef]
- Bresó-Femenia, E.; Chaudret, B.; Castillón, S. Selective catalytic hydrogenation of polycyclic aromatic hydrocarbons promoted by ruthenium Nanoparticles. Catal. Sci. Technol. 2015, 5, 2741–2751. [Google Scholar] [CrossRef]
- Castaño, P.; Van Herk, D.; Kreutzer, M.; Moulijn, J.A.; Makkee, M. Kinetic and deactivation modelling of biphenyl liquid-phase hydrogenation over bimetallic Pt–Pd catalyst. Appl. Catal. B Environ. 2009, 88, 213–223. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Koklin, A.E.; Bogdan, V.I.; Kustov, L.M. Hydrogenation of biphenyl and isomeric terphenyls over a Pt-containing catalyst. Rus. Chem. Bull. 2017, 66, 1208–1212. [Google Scholar] [CrossRef]
- Kalenchuk, A.; Bogdan, V.; Dunaev, S.; Kustov, L. Influence of steric factors on reversible reactions of hydrogenation-dehydrogenation of polycyclic aromatic hydrocarbons on a Pt/C catalyst in hydrogen storage systems. Fuel 2020, 280, 118625. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Bogdan, V.I.; Dunaev, S.F.; Kustov, L.M. Dehydrogenation of polycyclic naphthenes on a Pt/C catalyst for hydrogen storage in liquid organic hydrogen carriers. Fuel Process. Technol. 2018, 169, 94–100. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Kustov, L.M. Kinetic Modeling of Hydrogen Production by Dehydrogenation of Polycyclic Naphthenes with Varying Degrees of Condensation. Molecules 2022, 27, 2236. [Google Scholar] [CrossRef]
- Kustov, L.M.; Kalenchuk, A.N.; Dunaev, S.F.; Bogdan, V.I. Effect of isomerization on the performance of aromatic hydrogen storage systems possessing different condensation extents. Mendeleev Commun. 2019, 29, 25–28. [Google Scholar] [CrossRef]
- Kalenchuk, N.; Bogdan, V.I.; Dunaev, S.; Kustov, L. Effect of Isomerization on the Reversible Reaction of Hydrogenation-Dehydrogenation of ortho-Terphenyl on a Pt/C Catalyst. Chem. Eng. Technol. 2018, 41, 1842–1846. [Google Scholar] [CrossRef]
- Parker, D.H.; Pettiette-Hall, C.L.; Li, Y.; McIver, R.T.; Hemminger, J.C. Kinetic study of the initial stages of dehydrogenation of cyclohexane on the Pt(111) surface. J. Phys. Chem. 1992, 96, 1888–1894. [Google Scholar] [CrossRef]
- Pettiette-Hall, C.; Land, D.P.; McIver, R.T.; Hemminger, J.C. Identification of multiple steps in the dehydrogenation of cyclic C6 hydrocarbons to benzene on Pt(111). J. Am. Chem. Soc. 1991, 113, 2755–2766. [Google Scholar] [CrossRef]
- Herz, R.K.; Gillispie, W.D.; Petersen, E.E.; Somorjai, G.A. The structure sensitivity of cyclohexane dehydrogenation and hydrogenolysis catalyzed by platinum single crystals at atmospheric pressure. J. Catal. 1981, 67, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Karmakar, S.; Biniwale, R.B. Hydrogen delivery through liquid organic hydrides: Considerations for a potential technology. Int. J. Hydrog. Energy 2012, 37, 3719–3726. [Google Scholar] [CrossRef]


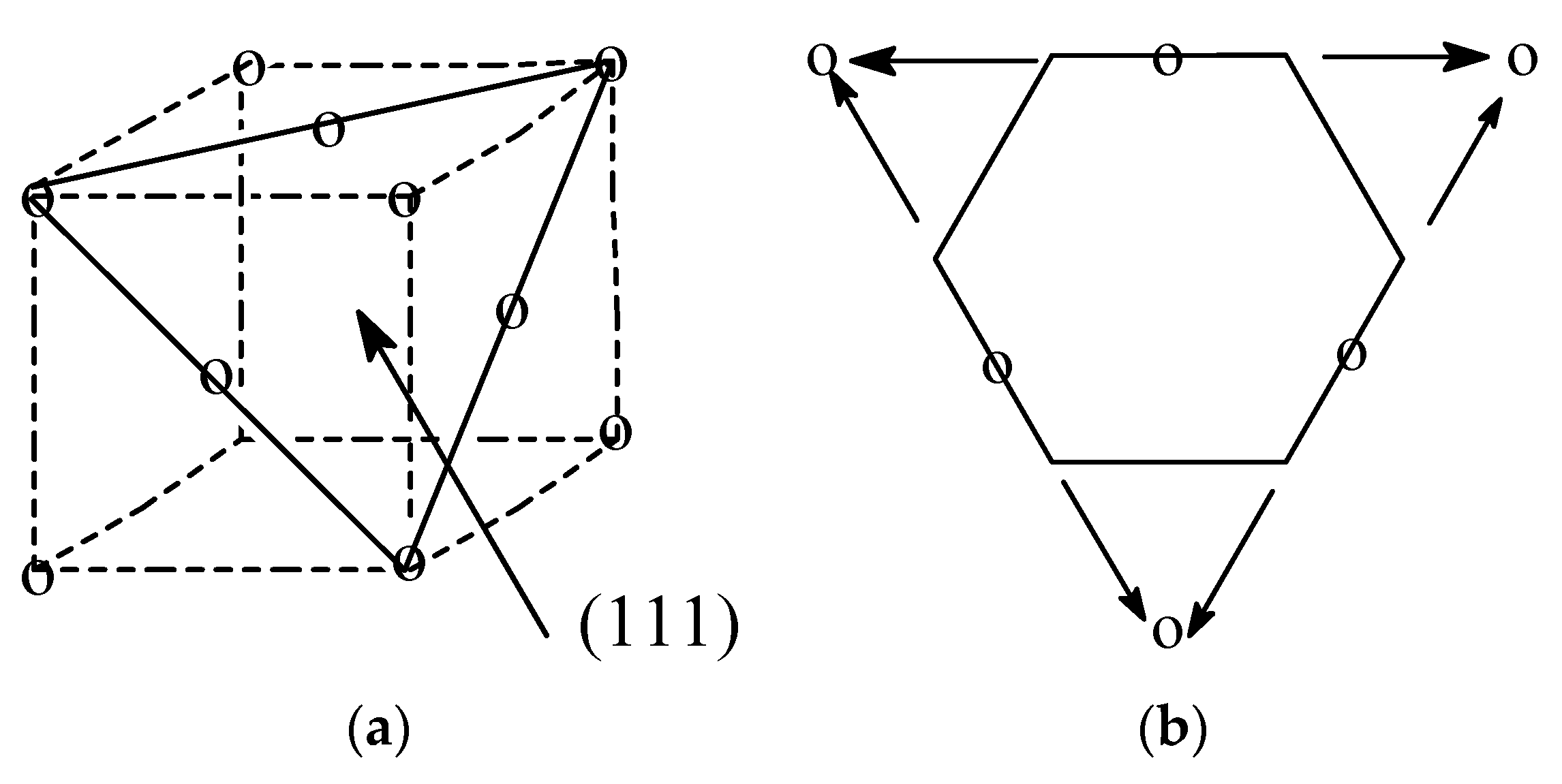
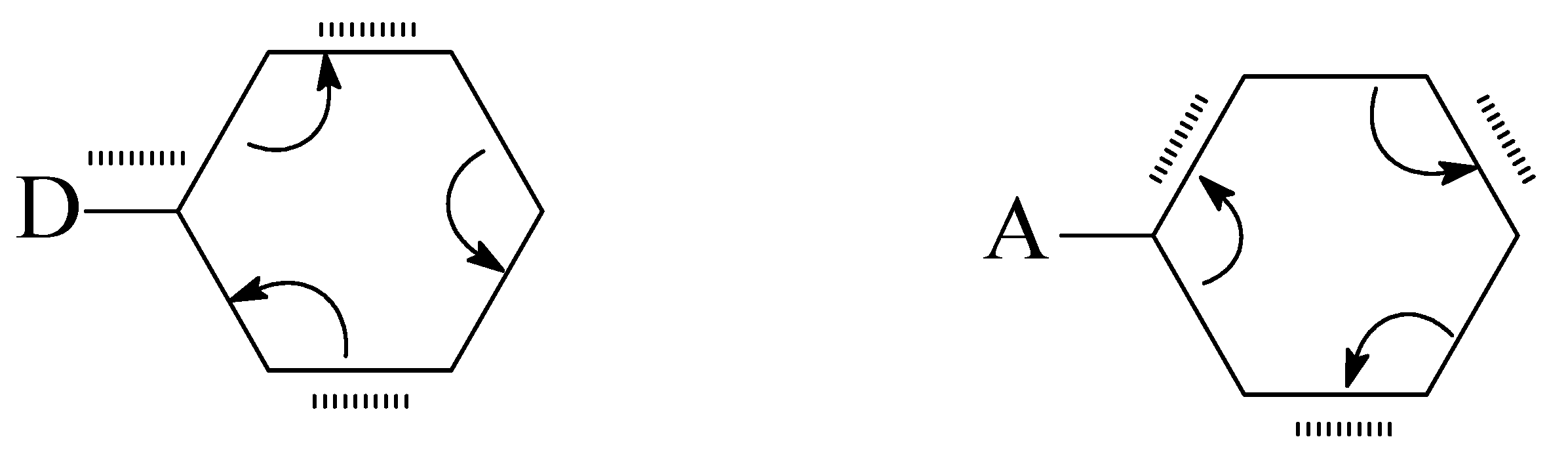

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Pt | Pd | Ir | Rh | Cu | Co | Ni |
|---|---|---|---|---|---|---|---|
| Parameter of the lattice, nm | 2.78 | 2.75 | 2.71 | 2.69 | 2.56 | 2.51 | 2.49 |
| Substrate | Catalyst | TOF (mmol(H2)/gPt × min) |
|---|---|---|
| Cyclohexane | 3.82 wt. % Pt/AC | 1800 |
| Methylcyclohexane | 3.82 wt. % Pt/AC | 1700 |
| Decalin | 3.82 wt. % Pt/C | 610 |
| Cyclohexane | 10 wt. % Ni/AC | 7.1 |
| Cyclohexane | 20 wt. % Ni/AC | 8.5 |
| Cyclohexane | 40 wt. % Pt/AC | 6.8 |
| Cyclohexane | 12 wt. % Pt-Rh/AC | 520 |
| Cyclohexane | 11 wt. % Pt-Re/AC | 550 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kustov, L.M.; Kalenchuk, A.N. The Role of the Metal in the Catalytic Reactions of Hydrogenation–Dehydrogenation of Polycyclic Hydrocarbons for Hydrogen Storage. Metals 2022, 12, 2002. https://doi.org/10.3390/met12122002
Kustov LM, Kalenchuk AN. The Role of the Metal in the Catalytic Reactions of Hydrogenation–Dehydrogenation of Polycyclic Hydrocarbons for Hydrogen Storage. Metals. 2022; 12(12):2002. https://doi.org/10.3390/met12122002
Chicago/Turabian StyleKustov, Leonid M., and Alexander N. Kalenchuk. 2022. "The Role of the Metal in the Catalytic Reactions of Hydrogenation–Dehydrogenation of Polycyclic Hydrocarbons for Hydrogen Storage" Metals 12, no. 12: 2002. https://doi.org/10.3390/met12122002
APA StyleKustov, L. M., & Kalenchuk, A. N. (2022). The Role of the Metal in the Catalytic Reactions of Hydrogenation–Dehydrogenation of Polycyclic Hydrocarbons for Hydrogen Storage. Metals, 12(12), 2002. https://doi.org/10.3390/met12122002