
In Vitro Degradation Behavior and Biocompatibility of Bioresorbable Molybdenum

Abstract
1. Introduction
2. Materials and Methods
2.1. Material Preparation
2.2. Electrolyte Preparation
2.3. Electrochemical Characterization
2.4. Static Immersion Test
2.5. Microstructural and Corrosion Product Characterization
2.6. Cell Culture
2.7. Time and Concentration Series
2.8. Determination of Mo Induced Apoptosis and Necrosis
2.9. Thrombogenicity Assay
2.10. Determination of Inflammation
2.11. RNA Isolation and Quantitative Real-Time PCR
2.12. Colonization of Metal Slices
2.13. Statistics Section
3. Results and Discussion
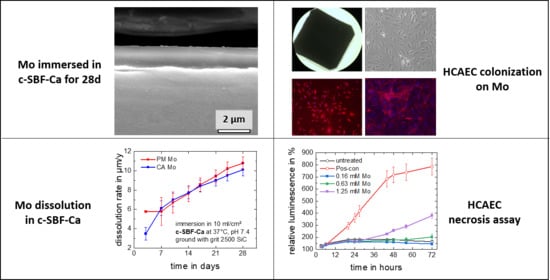
3.1. Degradation Properties
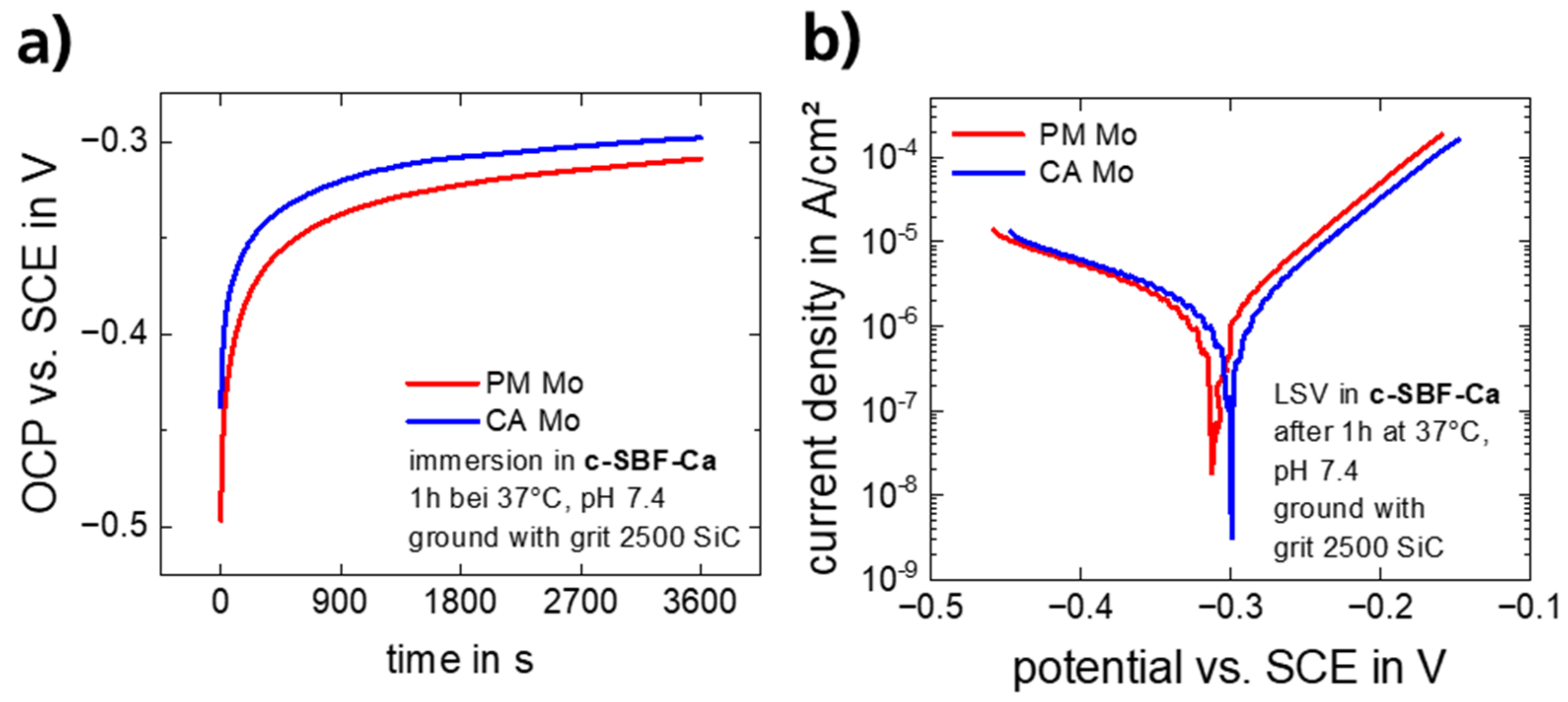
3.1.1. Electrochemical Corrosion Behavior
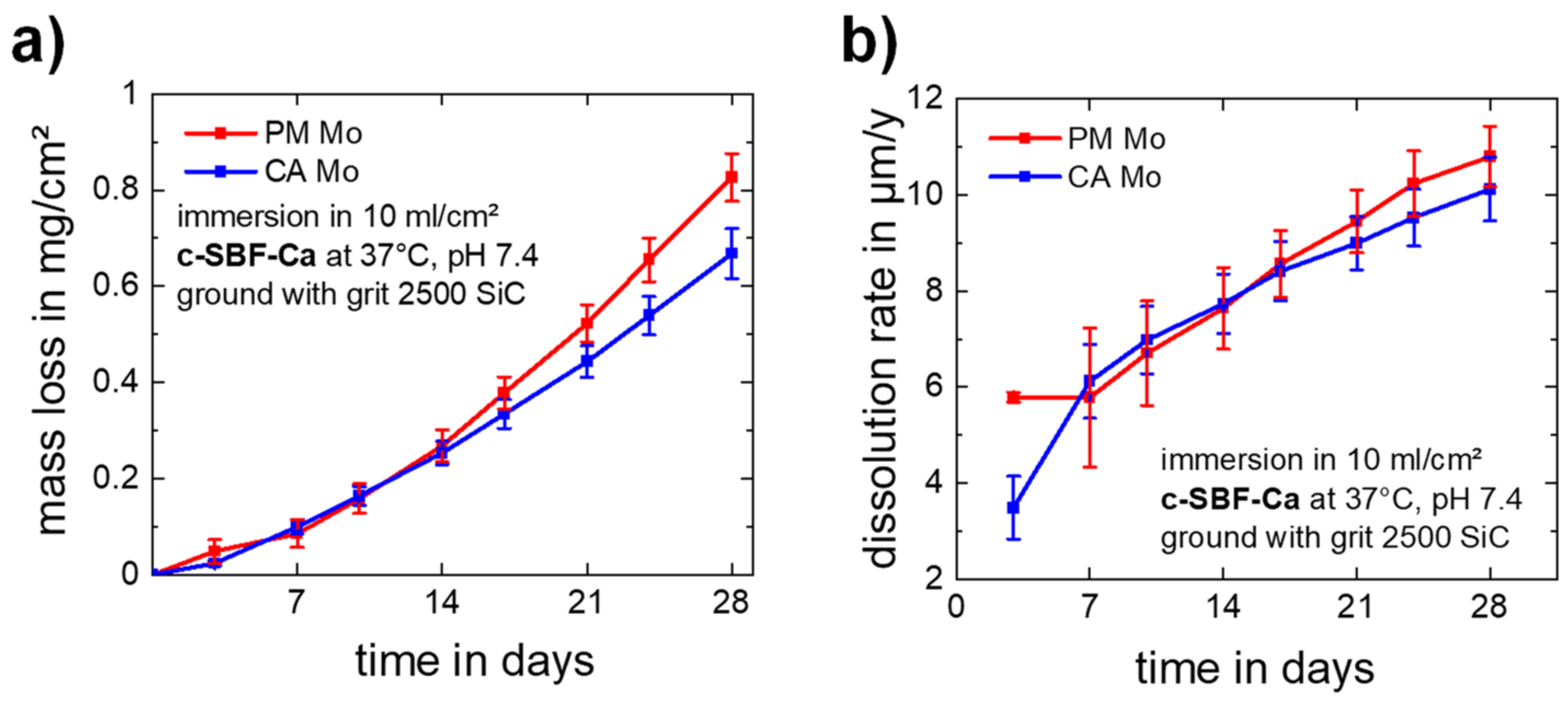
3.1.2. Static Immersion Corrosion Behavior
3.2. Biocompatibility
3.2.1. Dissolution Model for a Hypothetical Mo Stent
3.2.2. Cytotoxicity Time and Concentration Series
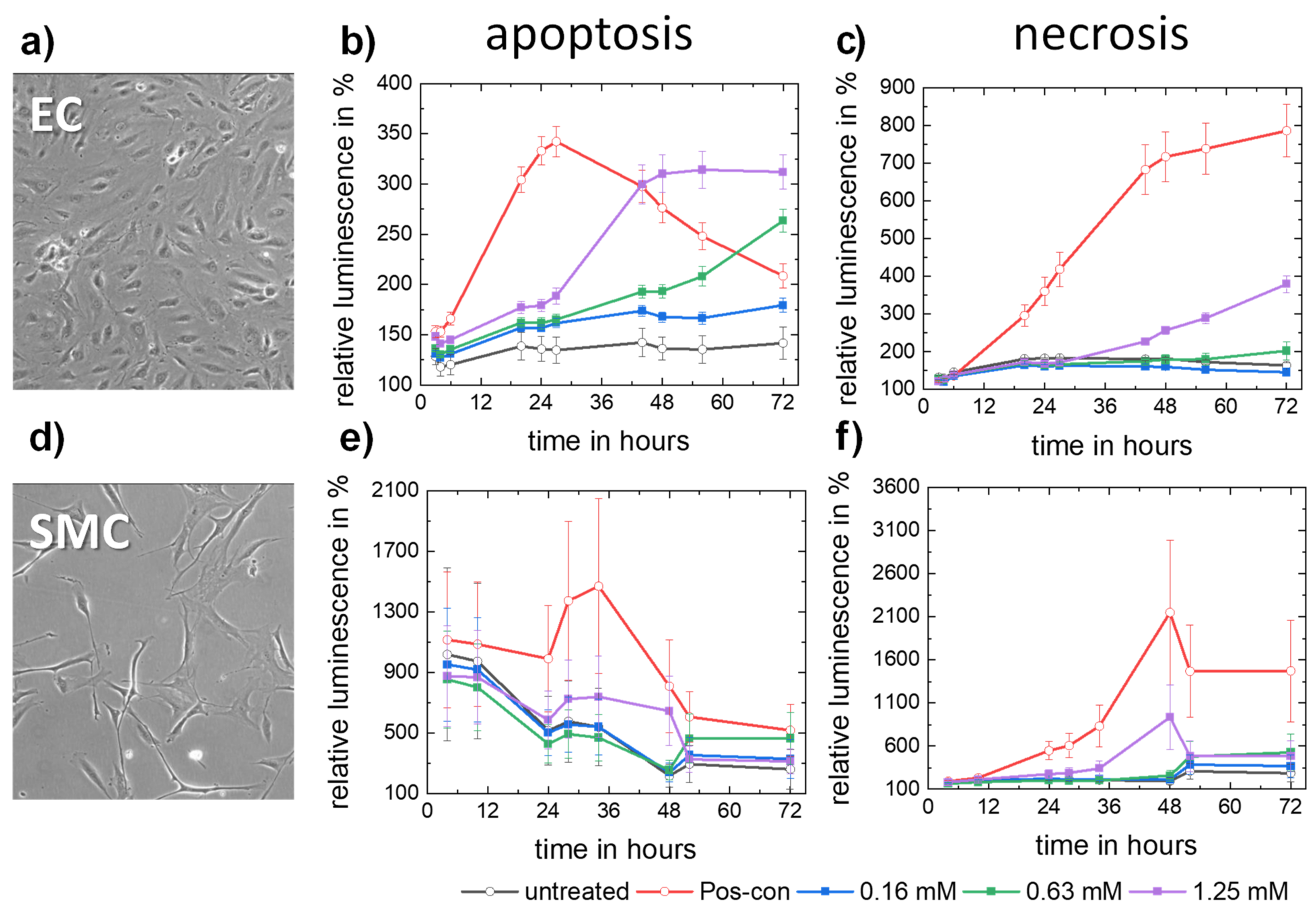
3.2.3. Apoptosis and Necrosis
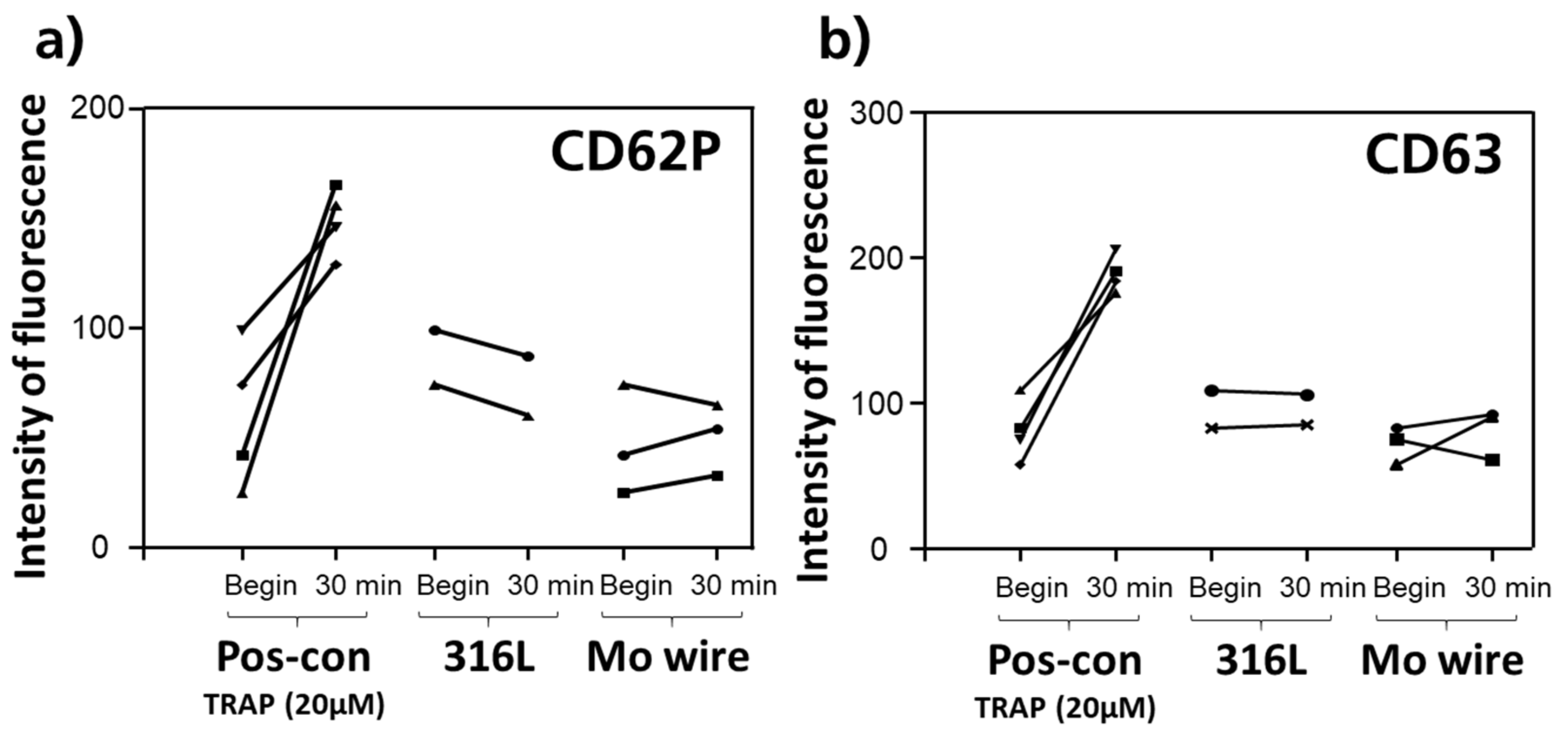
3.2.4. Thrombogenicity
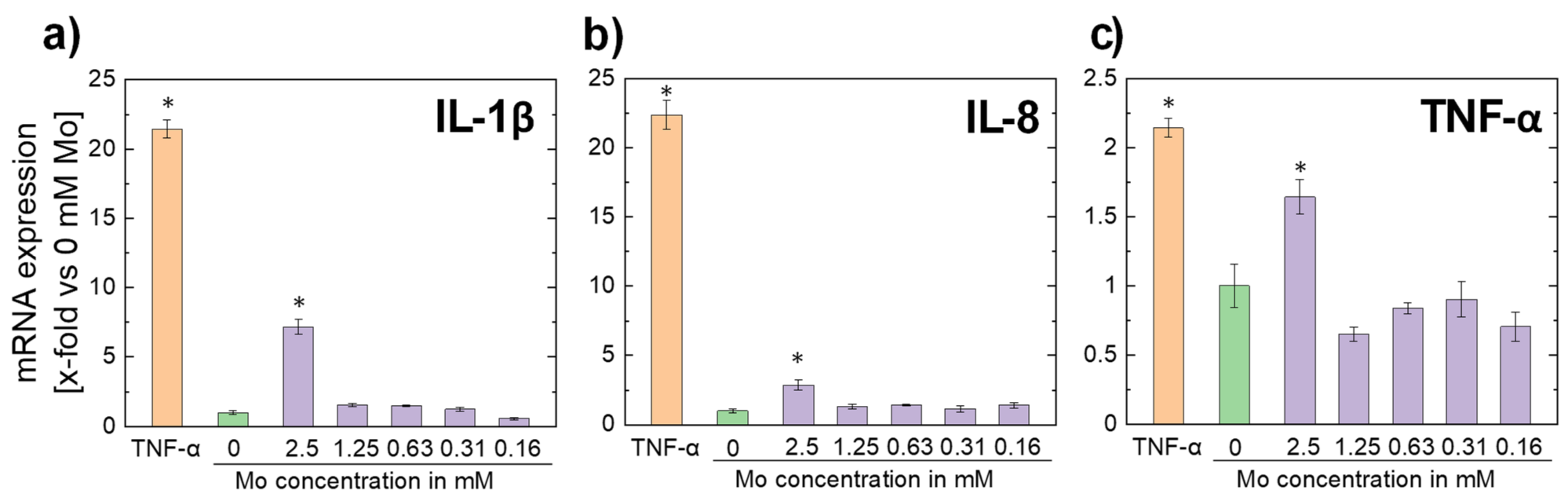
3.2.5. Inflammatory Response
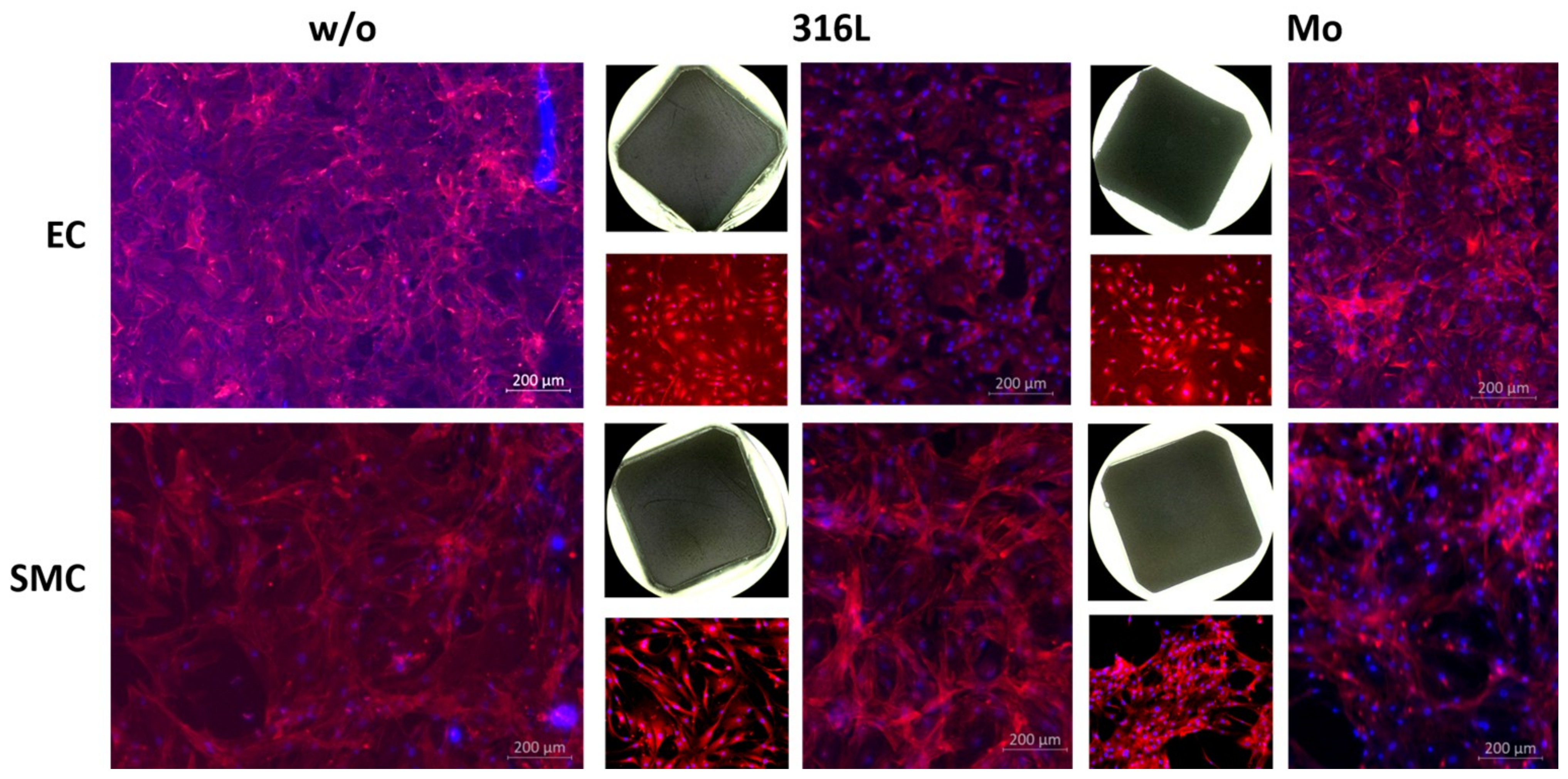
3.2.6. Colonization
4. Conclusions
- The corrosion attack on commercially available and powder metallurgically manufactured Mo in simulated physiological solution (c-SBF-Ca) is uniform over a period of 28 days and results in the formation of thin and non-passivating degradation product layers.
- Mo dissolution rates increase over time and reach a rate of approx. 10 µm/y after 28 days of immersion in c-SBF-Ca for both materials. This is the suitable order of magnitude for an application in stents.
- Molybdenum ion concentrations in the order of magnitude expected for a dissolving pure Mo stent do not trigger apoptosis or necrosis of human endothelial or smooth muscle cells.
- Molybdenum does not activate thrombocytes, i.e., no significant thrombogenicity is expected for a Mo implant.
- Molybdenum ion concentrations in the order of magnitude expected for a dissolving pure Mo stent do not trigger cytokine expression, i.e., an excessive immune reaction triggered by a Mo implant is unlikely.
- Molybdenum is colonizable by human endothelial and smooth muscle cells, i.e., a Mo stent might become well integrated into the (cardiovascular) vessel walls.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, Y.F.; Gu, X.N.; Witte, F. Biodegradable metals. Mater. Sci. Eng. R. Rep. 2014, 77, 1–34. [Google Scholar] [CrossRef]
- Ding, Y.; Wen, C.; Hodgson, P.; Li, Y. Effects of alloying elements on the corrosion behavior and biocompatibility of biodegradable magnesium alloys: A review. J. Mater. Chem. B 2014, 2, 1912–1933. [Google Scholar] [CrossRef]
- Wang, J.-L.; Xu, J.-K.; Hopkins, C.; Chow, D.H.-K.; Qin, L. Biodegradable Magnesium-Based Implants in Orthopedics-A General Review and Perspectives. Adv. Sci. 2020, 7, 1902443. [Google Scholar] [CrossRef]
- Jafari, S.; Harandi, S.E.; Singh Raman, R.K. A Review of Stress-Corrosion Cracking and Corrosion Fatigue of Magnesium Alloys for Biodegradable Implant Applications. JOM 2015, 67, 1143–1153. [Google Scholar] [CrossRef]
- Francis, A.; Yang, Y.; Virtanen, S.; Boccaccini, A.R. Iron and iron-based alloys for temporary cardiovascular applications. J. Mater. Sci. Mater. Med. 2015, 26. [Google Scholar] [CrossRef]
- Gorejová, R.; Haverová, L.; Oriňaková, R.; Oriňak, A.; Oriňak, M. Recent advancements in Fe-based biodegradable materials for bone repair. J. Mater. Sci. 2019, 54, 1913–1947. [Google Scholar] [CrossRef]
- Pierson, D.; Edick, J.; Tauscher, A.; Pokorney, E.; Bowen, P.; Gelbaugh, J.; Stinson, J.; Getty, H.; Lee, C.H.; Drelich, J.; et al. A simplified in vivo approach for evaluating the bioabsorbable behavior of candidate stent materials. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 58–67. [Google Scholar] [CrossRef]
- Kraus, T.; Moszner, F.; Fischerauer, S.; Fiedler, M.; Martinelli, E.; Eichler, J.; Witte, F.; Willbold, E.; Schinhammer, M.; Meischel, M.; et al. Biodegradable Fe-based alloys for use in osteosynthesis: Outcome of an in vivo study after 52 weeks. Acta Biomater. 2014, 10, 3346–3353. [Google Scholar] [CrossRef] [PubMed]
- Wegener, B.; Sichler, A.; Milz, S.; Sprecher, C.; Pieper, K.; Hermanns, W.; Jansson, V.; Nies, B.; Kieback, B.; Müller, P.E.; et al. Development of a novel biodegradable porous iron-based implant for bone replacement. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Bowen, P.K.; Drelich, J.; Goldman, J. Zinc exhibits ideal physiological corrosion behavior for bioabsorbable stents. Adv. Mater. 2013, 25, 2577–2582. [Google Scholar] [CrossRef] [PubMed]
- Venezuela, J.; Dargusch, M.S. The influence of alloying and fabrication techniques on the mechanical properties, biodegradability and biocompatibility of zinc: A comprehensive review. Acta Biomater. 2019, 87, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jia, B.; Zhang, Z.; Qu, X.; Li, G.; Lin, W.; Zhu, D.; Dai, K.; Zheng, Y. Alloying design of biodegradable zinc as promising bone implants for load-bearing applications. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Poncin, P.; Proft, J. Stent Tubing: Understanding the Desired Attributes. Mater. Proc. Med. Dev. Conf. 2003. [Google Scholar] [CrossRef]
- Leichtfried, G.; Schneibel, J.H.; Heilmaier, M. Ductility and impact resistance of powder-metallurgical molybdenum-rhenium alloys. Metal. Mat. Trans. A 2006, 37, 2955–2961. [Google Scholar] [CrossRef]
- Redlich, C.; Quadbeck, P.; Thieme, M.; Kieback, B. Molybdenum—A biodegradable implant material for structural applications? Acta Biomater. 2020, 104, 241–251. [Google Scholar] [CrossRef]
- Yin, L.; Cheng, H.; Mao, S.; Haasch, R.; Liu, Y.; Xie, X.; Hwang, S.-W.; Jain, H.; Kang, S.-K.; Su, Y.; et al. Dissolvable Metals for Transient Electronics. Adv. Funct. Mater. 2014, 24, 645–658. [Google Scholar] [CrossRef]
- Badawy, W.A.; Al-Kharafi, F.M. Corrosion and passivation behaviors of molybdenum in aqueous solutions of different pH. Electrochim. Acta 1998, 44, 693–702. [Google Scholar] [CrossRef]
- Shaldaev, V.S.; Malofeeva, A.N.; Davydov, A.D. Determination of corrosion rate of molybdenum, rhenium and their alloys in sodium chloride solution by the method of Tafel extrapolation. Russ. J. Electrochem. 2014, 50, 994–998. [Google Scholar] [CrossRef]
- De Rosa, L.; Tomachuk, C.R.; Springer, J.; Mitton, D.B.; Saiello, S.; Bellucci, F. The wet corrosion of molybdenum thin film–. Part I: Behavior at 25 °C. Mater. Corros. 2004, 55, 602–609. [Google Scholar] [CrossRef]
- Mörsdorf, A.; Odnevall Wallinder, I.; Hedberg, Y. Bioaccessibility of micron-sized powder particles of molybdenum metal, iron metal, molybdenum oxides and ferromolybdenum--Importance of surface oxides. Regul. Toxicol. Pharmacol. 2015, 72, 447–457. [Google Scholar] [CrossRef]
- Petrova, M.; Bojinov, M.; Zanna, S.; Marcus, P. Mechanism of anodic oxidation of molybdenum in nearly-neutral electrolytes studied by electrochemical impedance spectroscopy and X-ray photoelectron spectroscopy. Electrochim. Acta 2011, 56, 7899–7906. [Google Scholar] [CrossRef]
- Johnson, J.W.; Chi, C.H.; Chen, C.K.; James, W.J. The Anodic Dissolution of Molybdenum. Corrosion 1970, 26, 338–342. [Google Scholar] [CrossRef]
- Pourbaix, M. (Ed.) Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; NACE International: Houston, TX, USA, 1974; ISBN 0915567989. [Google Scholar]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef]
- Turnlund, J.R.; Keyes, W.R.; Peiffer, G.L.; Chiang, G. Molybdenum absorption, excretion, and retention studied with stable isotopes in young men during depletion and repletion. Am. J. Clin. Nutr. 1995, 61, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Kovalskii, V.V.; Iarovaia, G.A.; Shmavonyan, D.M. Modification of human and animal purine metabolism in conditions of various molybdenum bio-geochemical areas. Zhurnal Obs. Biol. 1961, 22, 179–191. [Google Scholar]
- Vyskočil, A.; Viau, C. Assessment of molybdenum toxicity in humans. J. Appl. Toxicol. 1999, 19, 185–192. [Google Scholar] [CrossRef]
- Molčilović, B. (Ed.) Dietary intakes of trace elements and related nutrients in eleven countries: Preliminary results from an International Atomic Energy Agency (IAEA) coordinated research programme. In Trace Elements in Man and Animals 7; Inst. for Medical Research and Occupational Health, University Zaqreb: Zagreb, Croatia, 1991; ISBN 8681477021. [Google Scholar]
- Caicedo, M.; Jacobs, J.J.; Reddy, A.; Hallab, N.J. Analysis of metal ion-induced DNA damage, apoptosis, and necrosis in human (Jurkat) T-cells demonstrates Ni2+ and V3+ are more toxic than other metals: Al3+, Be2+, Co2+, Cr3+, Cu2+, Fe3+, Mo5+, Nb5+, Zr2+. J. Biomed. Mater. Res. Part A 2008, 86, 905–913. [Google Scholar] [CrossRef]
- Daley, B.; Doherty, A.T.; Fairman, B.; Case, C.P. Wear debris from hip or knee replacements causes chromosomal damage in human cells in tissue culture. J. Bone Jt. Surg. 2004, 86, 598–606. [Google Scholar] [CrossRef]
- Okazaki, Y.; Rao, S.; Asao, S.; Tateishi, T. Effects of Metallic Concentrations Other Than Ti, Al and V on Cell Viability. Mater. Trans. JIM 1998, 39, 1070–1079. [Google Scholar] [CrossRef]
- Yamamoto, A.; Honma, R.; Sumita, M. Cytotoxicity evaluation of 43 metal salts using murine fibroblasts and osteoblastic cells. J. Biomed. Mater. Res. 1998, 39, 331–340. [Google Scholar] [CrossRef]
- Hallab, N.J.; Vermes, C.; Messina, C.; Roebuck, K.A.; Glant, T.T.; Jacobs, J.J. Concentration- and composition-dependent effects of metal ions on human MG-63 osteoblasts. J. Biomed. Mater. Res. 2002, 60, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- ASTM G31-72. Standard Practice for Laboratory Immersion Corrosion Testing of Metals; ASTM: West Conshohocken, PA, USA, 2004. [Google Scholar]
- Nikolaychuk, P.A.; Tyurin, A.G. The Revised Pourbaix Diagram for Molybdenum. Butl. Commun. 2011, 24, 101–105. [Google Scholar]
- Kumar, A.; Eyre, B.L. Grain boundary segregation and intergranular fracture in molybdenum. Proc. R. Soc. Lond. A 1980, 370, 431–458. [Google Scholar] [CrossRef]
- Al Doghaither, H.; Elmorsy, E.; Al-Ghafari, A.; Ghulam, J. Roles of oxidative stress, apoptosis, and inflammation in metal-induced dysfunction of beta pancreatic cells isolated from CD1 mice. Saudi J. Biol. Sci. 2021, 28, 651–663. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Al | Cr | Cu | Fe | Mn | Ni | Si | W | O | Mo |
|---|---|---|---|---|---|---|---|---|---|---|
| Mo powder | <50 | <100 | <50 | <30 | 2500 | Bal. | ||||
| CA Mo | <20 | <50 | <20 | <50 | <10 | <20 | <30 | Bal. | ||
| Mo wire | <10 | <20 | <20 | <20 | <10 | <20 | <300 | <40 | Bal. | |
| 316L powder | 17.72% | Bal. | 0.09% | 12.23% | 3400 | 2800 | 2.08% | |||
| 316L wire | 17.34% | 540 | Bal. | 1.86% | 13.68% | 4900 | 2.9% |
| Reagent [g/L] | Ion Concentration [mM] | ||
|---|---|---|---|
| NaCl | 7.996 | Na+ | 142.0 |
| NaHCO3 | 0.350 | K+ | 5.0 |
| KCl | 0.224 | Mg2+ | 1.5 |
| K H2PO4·3H2O | 0.228 | Ca2+ | 1.2 |
| MgCl2·6H2O | 0.305 | Cl− | 147.8 |
| CaCl2 | 0.139 | HCO3− | 4.2 |
| Na2SO4 | 0.071 | HPO42− | 1.0 |
| TRIS (CH2OH)3CNH2 | 6.057 | SO42− | 0.5 |
| 1 M HCl | 40 ml | ||
| Gene | Primer 1 | Primer 2 | Gene Bank ID |
|---|---|---|---|
| IL-1β | CAG TGG CAA TGA GGA TGA C | ATC GTG CAC ATA AGC CTC G | NM_000576 |
| IL-8 | GGA CAA GAG CCA GGA AGA AAC C | CTT CAA AAA CTT CTC CAC AAC | XM_031289 |
| TNF-α | GGCAGT CAG ATC ATC TTC TC | GAT AGA TGG GCT CAT ACC AG | NM_000594 |
| HPRT-1 | CTT TGC TGA CCT GCT GGA TTA C | TTG CGA CCT TGA CCA TCT TTG | NM_000194 |
| Material | OCP [mV] | Ekorr [mV] | ikorr [µA/cm2] | vkorr [µm/y] | ba [mV/dec] | bk [mV/dec] |
|---|---|---|---|---|---|---|
| CA Mo | −302 ± 2.7 | −306.3 ± 4.9 | 1.11 ± 0.31 | 5.7 ± 1.2 | 71.3 ± 1.5 | −149.0 ± 8.4 |
| PM Mo | −310.3 ± 1.2 | −312.3 ± 0.8 | 1.39 ± 0.04 | 7.1 ± 0.1 | 71.7 ± 1.5 | −139.6 ± 6.4 |
| wt. % | O | Na | Mg | P | Ca | Mo |
|---|---|---|---|---|---|---|
| 1 | 13.8 | 2.0 | 4.2 | 80.0 | ||
| 2 | 12.4 | 87.6 | ||||
| 3 | 10.7 | 89.3 | ||||
| 4 | 100.0 | |||||
| 5 | 19.8 | 0.9 | 0.8 | 3.3 | 6.7 | 68.5 |
| 6 | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redlich, C.; Schauer, A.; Scheibler, J.; Poehle, G.; Barthel, P.; Maennel, A.; Adams, V.; Weissgaerber, T.; Linke, A.; Quadbeck, P. In Vitro Degradation Behavior and Biocompatibility of Bioresorbable Molybdenum. Metals 2021, 11, 761. https://doi.org/10.3390/met11050761
Redlich C, Schauer A, Scheibler J, Poehle G, Barthel P, Maennel A, Adams V, Weissgaerber T, Linke A, Quadbeck P. In Vitro Degradation Behavior and Biocompatibility of Bioresorbable Molybdenum. Metals. 2021; 11(5):761. https://doi.org/10.3390/met11050761
Chicago/Turabian StyleRedlich, Christian, Antje Schauer, Jakob Scheibler, Georg Poehle, Peggy Barthel, Anita Maennel, Volker Adams, Thomas Weissgaerber, Axel Linke, and Peter Quadbeck. 2021. "In Vitro Degradation Behavior and Biocompatibility of Bioresorbable Molybdenum" Metals 11, no. 5: 761. https://doi.org/10.3390/met11050761
APA StyleRedlich, C., Schauer, A., Scheibler, J., Poehle, G., Barthel, P., Maennel, A., Adams, V., Weissgaerber, T., Linke, A., & Quadbeck, P. (2021). In Vitro Degradation Behavior and Biocompatibility of Bioresorbable Molybdenum. Metals, 11(5), 761. https://doi.org/10.3390/met11050761