

Epigenomic Echoes—Decoding Genomic and Epigenetic Instability to Distinguish Lung Cancer Types and Predict Relapse

 , , , and
, , , and 
Abstract
1. Introduction
1.1. Overview of Genomic Instability
1.1.1. Mechanisms Leading to Genomic Instability
1.1.2. Consequences of Genomic Instability
1.1.3. Genomic Instability as a Cancer Hallmark
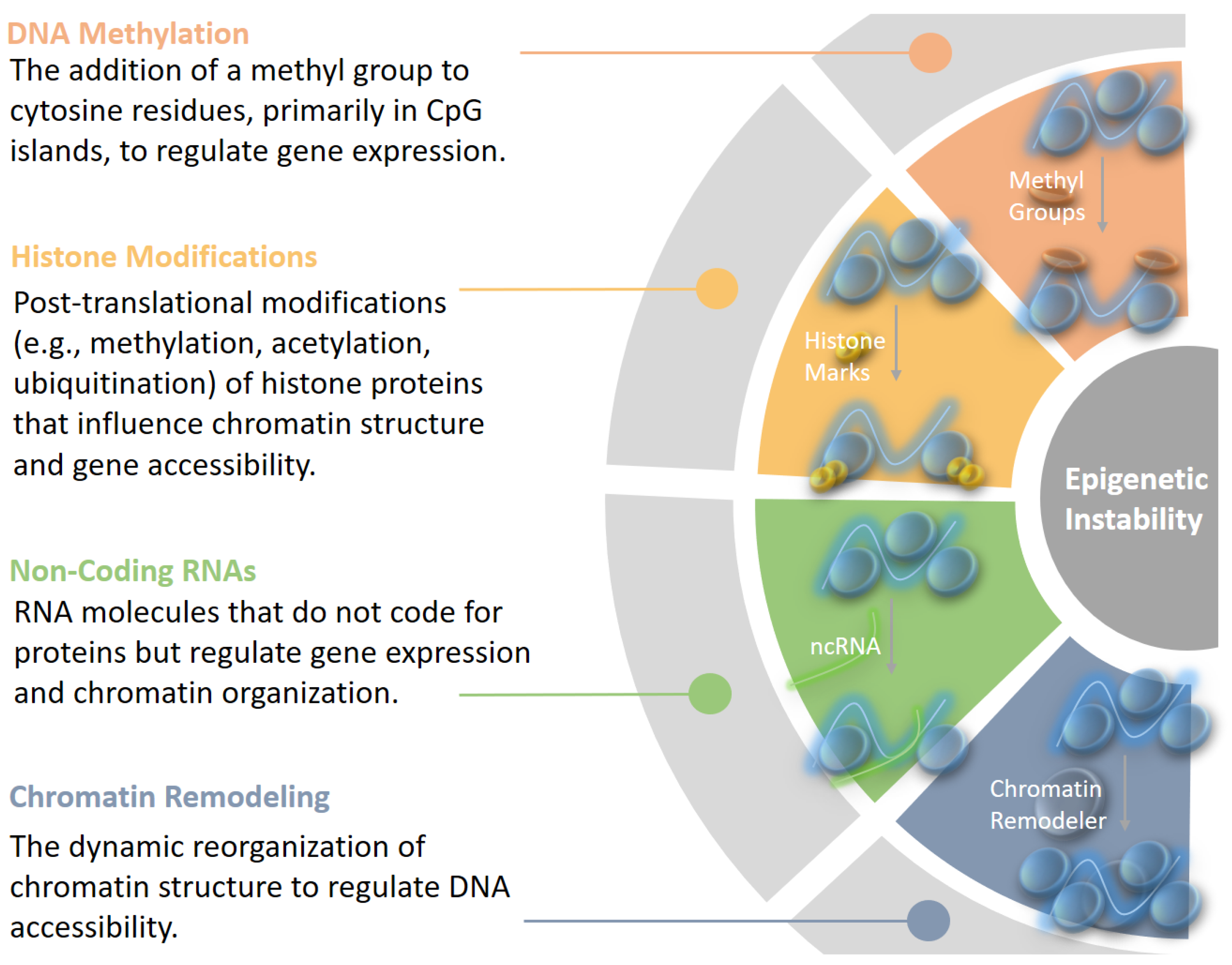
1.2. Overview of Epigenetic Instability
- Global Hypomethylation: This phenomenon involves the loss of methylation across the genome, which can lead to chromosomal instability and activate oncogenes. It is commonly observed in various cancers.
- Localized Hypermethylation: Specific regions, particularly the promoters of tumor suppressor genes, can undergo hypermethylation, resulting in gene silencing. This silencing removes critical checks on cell growth and division, thus enabling tumor growth.
- Disrupted Chromatin Accessibility: Aberrant remodeling can result in either hyper-compacted or excessively open chromatin, impairing transcription and DNA repair processes [36].
- Histone Modification Alterations: Mutations in remodelers (e.g., SWI/SNF components) disrupt interactions with histone-modifying enzymes, silencing tumor suppressor genes or activating oncogenes [37].
- Nucleosome Instability: Improper incorporation of histone variants destabilizes chromatin, increasing susceptibility to DNA damage [38].
- Compromised DNA Repair: Defective remodeling hinders repair pathways like homologous recombination, leading to the accumulation of mutations and further epigenetic changes [39].
2. Relationship Between Genomic and Epigenetic Instability
2.1. Epigenetic Crosstalk Between Mountains and Valleys
2.2. Epigenetic Regulation of DNA Repair Genes
2.3. Implications for Tumor Evolution and Heterogeneity
3. Epigenetic Biomarkers for Lung Cancer Type Distinction and Relapse
3.1. Biomarker Identification via Epigenetic Marks
3.2. Practical Classification and Screening Applications in Oncology
3.3. Epigenetic Indicators of Cancer Relapse
4. Advancing Therapeutic Strategies—Next Steps and Unmet Needs
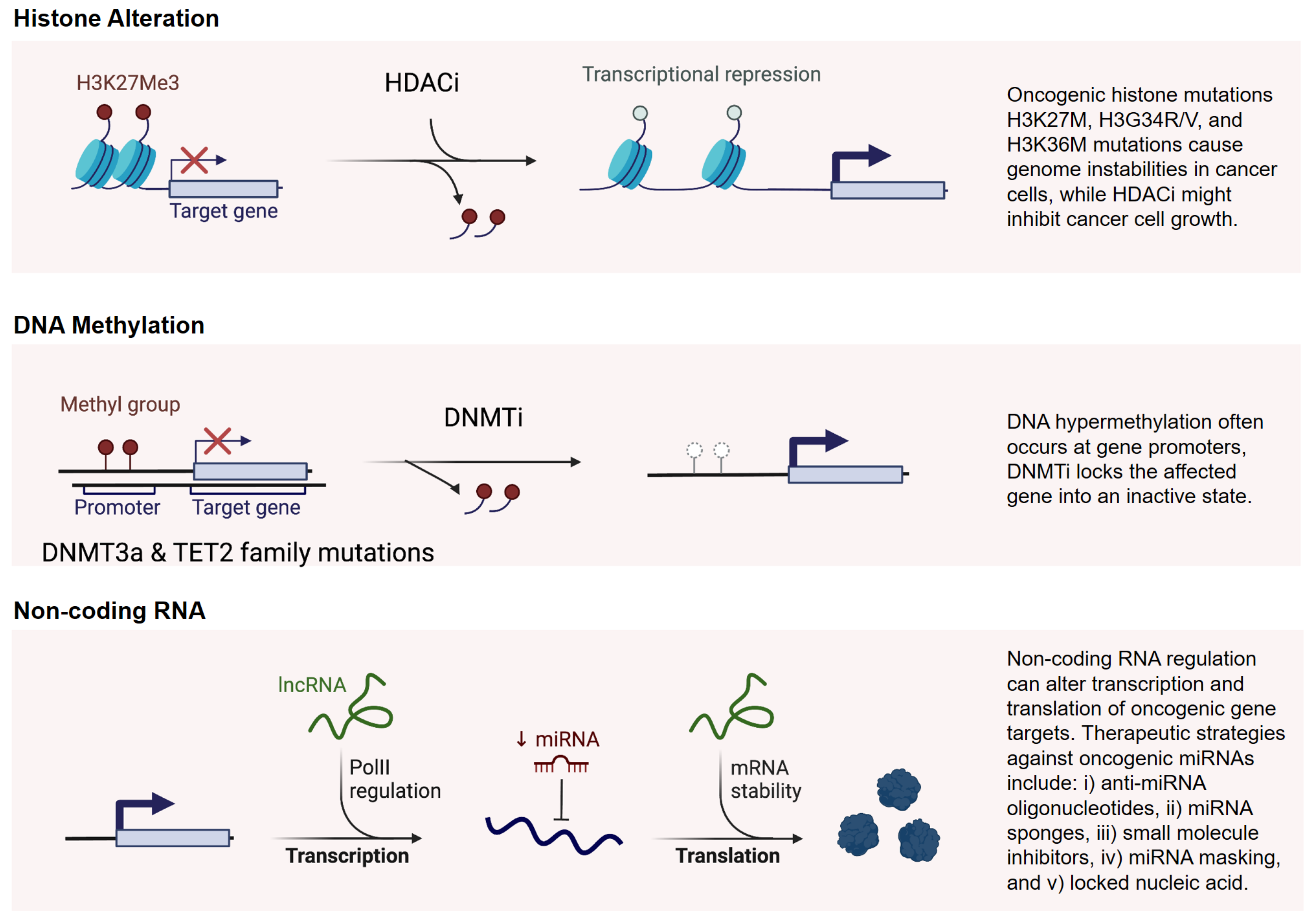
4.1. Targeting Genomic Instability
4.2. Targeting Epigenetic Modifications
4.3. Natural Epi-Drugs as Emerging Tools for Epigenetic Modulation
4.4. Ethics of Implementing Epigenomics Technologies in Cancer Screening and Treatment
4.5. Future Directions—Multi-Omics Integration for Comprehensive Cancer Therapy?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Rawi, D.H.; Bakhoum, S.F. Chromosomal instability as a source of genomic plasticity. Curr. Opin. Genet. Dev. 2022, 74, 101913. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Ried, T.; Meijer, G.A.; Harrison, D.J.; Grech, G.; Franch-Expósito, S.; Briffa, R.; Carvalho, B.; Camps, J. The landscape of genomic copy number alterations in colorectal cancer and their consequences on gene expression levels and disease outcome. Mol. Asp. Med. 2019, 69, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Cleal, K.; Baird, D.M. Catastrophic Endgames: Emerging Mechanisms of Telomere-Driven Genomic Instability. Trends Genet. Tig 2020, 36, 347–359. [Google Scholar] [CrossRef]
- Irony-Tur Sinai, M.; Kerem, B. Genomic instability in fragile sites—still adding the pieces. Genes, Chromosom. Cancer 2019, 58, 295–304. [Google Scholar] [CrossRef]
- López-Gil, L.; Pascual-Ahuir, A.; Proft, M. Genomic Instability and Epigenetic Changes during Aging. Int. J. Mol. Sci. 2023, 24, 14279. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res. 2017, 77, 2179. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620. [Google Scholar] [CrossRef]
- Siri, S.O.; Martino, J.; Gottifredi, V. Structural Chromosome Instability: Types, Origins, Consequences, and Therapeutic Opportunities. Cancers 2021, 13, 3056. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell 2023, 186, 528–542.e14. [Google Scholar] [CrossRef]
- Trerotola, M.; Relli, V.; Simeone, P.; Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genom. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Jain, R.; Yadav, R.K. Emerging roles of cancer-associated histone mutations in genomic instabilities. Front. Cell Dev. Biol. 2024, 12, 1455572. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [PubMed]
- Naselli, F.; Cardinale, P.S.; Volpes, S.; Martino, C.; Cruciata, I.; Valenti, R.; Luparello, C.; Caradonna, F.; Chiarelli, R. An alternative approach of TUNEL assay to specifically characterize DNA fragmentation in cell model systems. Histochem. Cell Biol. 2024, 162, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297. [Google Scholar] [CrossRef] [PubMed]
- Galassi, C.; Chan, T.A.; Vitale, I.; Galluzzi, L. The hallmarks of cancer immune evasion. Cancer Cell 2024, 42, 1825–1863. [Google Scholar] [CrossRef] [PubMed]
- WHO. Lung Cancer. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/lung-cancer (accessed on 1 February 2025).
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, H.; Xu, J. Construction of a genomic instability-derived predictive prognostic signature for non-small cell lung cancer patients. Cancer Genet. 2023, 278–279, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Tian, T.; Pang, L. Mathematical modelling the pathway of genomic instability in lung cancer. Sci. Rep. 2019, 9, 14136. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhao, E.; Yu, H.; Yuan, C.; Abbas, M.N.; Cui, H. Methylation across the central dogma in health and diseases: New therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 310. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, M.; Wang, Y. The roles of histone modifications in tumorigenesis and associated inhibitors in cancer therapy. J. Natl. Cancer Cent. 2022, 2, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, F.L.; Cheng, Z.L.; Lei, Q.Y. Impact of acetylation on tumor metabolism. Mol. Cell. Oncol. 2014, 1, e963452. [Google Scholar] [CrossRef] [PubMed]
- Nimal, S.; Kumbhar, N.; Saruchi; Rathore, S.; Naik, N.; Paymal, S.; Gacche, R.N. Apigenin and its combination with Vorinostat induces apoptotic-mediated cell death in TNBC by modulating the epigenetic and apoptotic regulators and related miRNAs. Sci. Rep. 2024, 14, 9540. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Uppaluri, K.R.; Challa, H.J.; Gaur, A.; Jain, R.; Krishna Vardhani, K.; Geddam, A.; Natya, K.; Aswini, K.; Palasamudram, K.; K, S.M. Unlocking the potential of non-coding RNAs in cancer research and therapy. Transl. Oncol. 2023, 35, 101730. [Google Scholar] [CrossRef]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Q.; Jiang, W.; Bian, Y.; Zhou, Y.; Gou, A.; Zhang, W.; Fu, K.; Shi, W. Emerging roles of piRNAs in cancer: Challenges and prospects. Aging 2019, 11, 9932. [Google Scholar] [CrossRef]
- Ponzetti, M.; Rucci, N.; Falone, S. RNA methylation and cellular response to oxidative stress-promoting anticancer agents. Cell Cycle 2023, 22, 870. [Google Scholar] [CrossRef] [PubMed]
- Sendinc, E.; Shi, Y. RNA m6A methylation across the transcriptome. Mol. Cell 2023, 83, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, R.; Li, Y.; Jiang, L.; Ma, D.; Luo, Q.; Song, G. Chromatin accessibility: Biological functions, molecular mechanisms and therapeutic application. Signal Transduct. Target. Ther. 2024, 9, 340. [Google Scholar] [CrossRef]
- Downs, J.A.; Gasser, S.M. Chromatin remodeling and spatial concerns in DNA double-strand break repair. Curr. Opin. Cell Biol. 2024, 90, 102405. [Google Scholar] [CrossRef]
- Orlando, K.A.; Nguyen, V.; Raab, J.R.; Walhart, T.; Weissman, B.E. Remodeling the cancer epigenome: Mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev. Anticancer. Ther. 2019, 19, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, D.; Vagnarelli, P. Contribution of histone variants to aneuploidy: A cancer perspective. Front. Genet. 2023, 14. [Google Scholar] [CrossRef]
- Fernandez, A.; O’Leary, C.; O’Byrne, K.J.; Burgess, J.; Richard, D.J.; Suraweera, A. Epigenetic Mechanisms in DNA Double Strand Break Repair: A Clinical Review. Front. Mol. Biosci. 2021, 8, 685440. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Bigio, M.R.D.; Rastegar, M.; Davie, J.R. DNA Modifications: Function and Applications in Normal and Disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, D.; Inaba, S.; Kitagawa, Y.; Tsukakoshi, K.; Ikebukuro, K. CpG Methylation Altered the Stability and Structure of the i-Motifs Located in the CpG Islands. Int. J. Mol. Sci. 2022, 23, 6467. [Google Scholar] [CrossRef]
- Lorenzatti, A.; Piga, E.J.; Gismondi, M.; Binolfi, A.; Margarit, E.; Calcaterra, N.B.; Armas, P. Genetic variations in G-quadruplex forming sequences affect the transcription of human disease-related genes. Nucleic Acids Res. 2023, 51, 12124. [Google Scholar] [CrossRef]
- Kuo, F.C.; Neville, M.J.; Sabaratnam, R.; Wesolowska-Andersen, A.; Phillips, D.; Wittemans, L.B.; van Dam, A.D.; Loh, N.Y.; Todorčević, M.; Denton, N.; et al. HOTAIR interacts with PRC2 complex regulating the regional preadipocyte transcriptome and human fat distribution. Cell Rep. 2022, 40, 111136. [Google Scholar] [CrossRef]
- Szczepanek, J.; Tretyn, A. MicroRNA-Mediated Regulation of Histone-Modifying Enzymes in Cancer: Mechanisms and Therapeutic Implications. Biomolecules 2023, 13, 1590. [Google Scholar] [CrossRef]
- Ma, B.; Wang, S.; Wu, W.; Shan, P.; Chen, Y.; Meng, J.; Xing, L.; Yun, J.; Hao, L.; Wang, X.; et al. Mechanisms of circRNA/lncRNA-miRNA interactions and applications in disease and drug research. Biomed. Pharmacother. 2023, 162, 114672. [Google Scholar] [CrossRef]
- Babayev, M.; Silveyra, P. Role of circular RNAs in lung cancer. Front. Genet. 2024, 15, 1346119. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Lange, T.d. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.; Qamra, A.; Chu, T.W.; Sharma, M.; Tsao, J.; Henry, D.A.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Winegarden, N.; Lupien, M.; et al. Telomere dysfunction cooperates with epigenetic alterations to impair murine embryonic stem cell fate commitment. eLife 2020, 9, e47333. [Google Scholar] [CrossRef]
- Rivosecchi, J.; Jurikova, K.; Cusanelli, E. Telomere-specific regulation of TERRA and its impact on telomere stability. Semin. Cell Dev. Biol. 2024, 157, 3–23. [Google Scholar] [CrossRef]
- Rossi, M.; Gorospe, M. Noncoding RNAs controlling telomere homeostasis in senescence and aging. Trends Mol. Med. 2020, 26, 422–433. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA Methylation in the Resistance to Therapy in Solid Tumors. Front. Oncol. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 2011, 19, 86. [Google Scholar] [CrossRef]
- Sun, S.; Yu, F.; Xu, D.; Zheng, H.; Li, M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim. Biophys. Acta-(BBA)-Rev. Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef]
- Morrison, A.J. Chromatin-remodeling links metabolic signaling to gene expression. Mol. Metab. 2020, 38, 100973. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Sood, S.; Dykhuizen, E.C. At the Crossroad of Gene Regulation and Genome Organization: Potential Roles for ATP-Dependent Chromatin Remodelers in the Regulation of CTCF-Mediated 3D Architecture. Biology 2021, 10, 272. [Google Scholar] [CrossRef]
- Herr, L.M.; Schaffer, E.D.; Fuchs, K.F.; Datta, A.; Brosh, R.M. Replication stress as a driver of cellular senescence and aging. Commun. Biol. 2024, 7, 616. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Fu, Y.; Li, X. Roles of post-translational modifications of UHRF1 in cancer. Epigenetics Chromatin 2024, 17, 15. [Google Scholar] [CrossRef]
- Chen, J.F.; Yan, Q. The roles of epigenetics in cancer progression and metastasis. Biochem. J. 2021, 478, 3373. [Google Scholar] [CrossRef]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Castro, M.d.C.C.; Deese, A.R.; Zhang, L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef] [PubMed]
- Terekhanova, N.V.; Karpova, A.; Liang, W.W.; Strzalkowski, A.; Chen, S.; Li, Y.; Southard-Smith, A.N.; Iglesia, M.D.; Wendl, M.C.; Jayasinghe, R.G.; et al. Epigenetic regulation during cancer transitions across 11 tumour types. Nature 2023, 623, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Du, H.; Li, B.; Luo, Z.; Zhu, L. Unlocking phenotypic plasticity provides novel insights for immunity and personalized therapy in lung adenocarcinoma. Front. Genet. 2022, 13, 941567. [Google Scholar] [CrossRef]
- Chen, J.; Pan, X.; Na, F.; Chen, X.; Chen, C. Epigenetic reprogramming in small cell lung cancer. Cancer Biol. Med. 2022, 19, 1111. [Google Scholar] [CrossRef] [PubMed]
- Solta, A.; Ernhofer, B.; Boettiger, K.; Megyesfalvi, Z.; Heeke, S.; Hoda, M.A.; Lang, C.; Aigner, C.; Hirsch, F.R.; Schelch, K.; et al. Small cells–big issues: Biological implications and preclinical advancements in small cell lung cancer. Mol. Cancer 2024, 23, 41. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.M.; Kartha, V.K.; Ma, S.; Meli, K.; Del Priore, I.; Lareau, C.; Naranjo, S.; Westcott, P.M.K.; Duarte, F.M.; Sankar, V.; et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020, 38, 212–228.e13. [Google Scholar] [CrossRef] [PubMed]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer 2009, 124, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, R.; Zhang, X.; Sun, Y.; Liu, P.; Francoeur, N.; Han, L.; Lam, W.Y.; Yi, Z.; Sebra, R.; et al. LINE-1 promotes tumorigenicity and exacerbates tumor progression via stimulating metabolism reprogramming in non-small cell lung cancer. Mol. Cancer 2022, 21, 147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684. [Google Scholar] [CrossRef]
- Garcia-Venzor, A.; Toiber, D. SIRT6 Through the Brain Evolution, Development, and Aging. Front. Aging Neurosci. 2021, 13, 747989. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Qian, M.; Peng, B.; Peng, L.; Wang, X.; Zheng, K.; Liu, Z.; Tang, X.; Zhang, S.; Sun, S.; et al. Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. eLife 2020, 9, e55828. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, Y.; Song, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Z.; Wang, Y. The dual role of sirtuins in cancer: Biological functions and implications. Front. Oncol. 2024, 14, 1384928. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Swarnkar, S.; Bhaskar, L.; Verma, H.K. Non-Coding RNA as a Biomarker in Lung Cancer. Non-Coding RNA 2024, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bian, T.; She, B.; Liu, L.; Sun, H.; Zhang, Q.; Zhu, J.; Zhang, J.; Liu, Y. Evaluating the comprehensive diagnosis efficiency of lung cancer, including measurement of SHOX2 and RASSF1A gene methylation. BMC Cancer 2024, 24, 282. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, X.; Li, H.; Yin, T.; Jiang, Y. Prognosis of immunotherapy for non-small cell lung cancer with CDKN2A loss of function. J. Thorac. Dis. 2024, 16, 507–515. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Liu, F.; Xu, L.; Peng, F.; Zhao, N.; Fu, B.; Zhu, Z.; Shi, Y.; Liu, J.; et al. A systematic review and meta-analysis: Association between MGMT hypermethylation and the clinicopathological characteristics of non-small-cell lung carcinoma. Sci. Rep. 2018, 8, 1439. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, H.; Kim, H.; Shim, Y.M.; Han, J.; Park, J.; Kim, D.H. Promoter methylation of retinoic acid receptor beta 2 and the development of second primary lung cancers in non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, X.; Zhou, X.; Huang, H. APC gene promoter methylation as a potential biomarker for lung cancer diagnosis: A meta-analysis. Thorac. Cancer 2021, 12, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Mirdamadi, M.S.A.; Talebi, Y.; Khaniabad, N.; Banaei, G.; Daneii, P.; Gholami, S.; Ghorbani, A.; Tavakolpournegari, A.; Farsani, Z.M.; et al. Pre-clinical and clinical importance of miR-21 in human cancers: Tumorigenesis, therapy response, delivery approaches and targeting agents. Pharmacol. Res. 2023, 187, 106568. [Google Scholar] [CrossRef]
- Kalfert, D.; Ludvikova, M.; Pesta, M.; Ludvik, J.; Dostalova, L.; Kholová, I. Multifunctional Roles of miR-34a in Cancer: A Review with the Emphasis on Head and Neck Squamous Cell Carcinoma and Thyroid Cancer with Clinical Implications. Diagnostics 2020, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lu, W.; Xu, J.; Shi, Y.; Zhang, H.; Xia, D. Prognostic value of long non-coding RNA MALAT1 in cancer patients. Tumor Biol. 2016, 37, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Zhu, Q.N.; Zhang, H.B.; Hu, Y.; Wang, G.; Zhu, Y.S. MALAT1: A potential biomarker in cancer. Cancer Manag. Res. 2018, 10, 6757–6768. [Google Scholar] [CrossRef]
- Onishi, T.; Takashima, T.; Kurashige, M.; Ohshima, K.; Morii, E. Mutually exclusive expression of EZH2 and H3K27me3 in non-small cell lung carcinoma. Pathol.-Res. Pract. 2022, 238, 154071. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E. Detection and relevance of epigenetic markers on ctDNA: Recent advances and future outlook. Mol. Oncol. 2021, 15, 1683–1700. [Google Scholar] [CrossRef]
- Chen, S.; Huang, K.; Zou, L.; Chen, L.; Hu, P. Diagnostic value of SHOX2, RASSF1A gene methylation combined with CEA level detection in malignant pleural effusion. BMC Pulm. Med. 2023, 23, 160. [Google Scholar] [CrossRef]
- Ibrahim, J.; Peeters, M.; Van Camp, G.; Op de Beeck, K. Methylation biomarkers for early cancer detection and diagnosis: Current and future perspectives. Eur. J. Cancer 2023, 178, 91–113. [Google Scholar] [CrossRef]
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Shin, S.H.; Kim, H.H.; Min, N.Y.; Lim, Y.; Joo, T.w.; Lee, K.J.; Jeong, M.S.; Kim, H.; Yun, S.y.; et al. Advances in methylation analysis of liquid biopsy in early cancer detection of colorectal and lung cancer. Sci. Rep. 2023, 13, 13502. [Google Scholar] [CrossRef]
- Khodadadi, E.; Fahmideh, L.; Khodadadi, E.; Dao, S.; Yousefi, M.; Taghizadeh, S.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Current Advances in DNA Methylation Analysis Methods. BioMed Res. Int. 2021, 2021, 8827516. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997, Erratum in Biology 2024, 13, 286. [Google Scholar] [CrossRef]
- Alexandrou, G.; Mantikas, K.T.; Allsopp, R.; Yapeter, C.A.; Jahin, M.; Melnick, T.; Ali, S.; Coombes, R.C.; Toumazou, C.; Shaw, J.A.; et al. The Evolution of Affordable Technologies in Liquid Biopsy Diagnostics: The Key to Clinical Implementation. Cancers 2023, 15, 5434. [Google Scholar] [CrossRef]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef]
- Guidry, K.; Vasudevaraja, V.; Labbe, K.; Mohamed, H.; Serrano, J.; Guidry, B.W.; DeLorenzo, M.; Zhang, H.; Deng, J.; Sahu, S.; et al. DNA Methylation Profiling Identifies Subgroups of Lung Adenocarcinoma with Distinct Immune Cell Composition, DNA Methylation Age, and Clinical Outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 3824–3835. [Google Scholar] [CrossRef] [PubMed]
- Heeke, S.; Gay, C.M.; Estecio, M.R.; Tran, H.; Morris, B.B.; Zhang, B.; Tang, X.; Raso, M.G.; Rocha, P.; Lai, S.; et al. Tumor- and circulating-free DNA methylation identifies clinically relevant small cell lung cancer subtypes. Cancer Cell 2024, 42, 225. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wu, Y. A prognosis-related molecular subtype for early-stage non-small lung cell carcinoma by multi-omics integration analysis. BMC Cancer 2021, 21, 128. [Google Scholar] [CrossRef]
- Xu, W.; Yao, H.; Wu, Z.; Yan, X.; Jiao, Z.; Liu, Y.; Zhang, M.; Wang, D. Oncoprotein SET-associated transcription factor ZBTB11 triggers lung cancer metastasis. Nat. Commun. 2024, 15, 1362. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, N.; Shulgin, B.; Katuninks, N.; Kulesh, V.; Helmlinger, G.; Zhudenkov, K.; Peskov, K. Assessment of NSCLC disease burden: A survival model-based meta-analysis study. Comput. Struct. Biotechnol. J. 2024, 24, 611–621. [Google Scholar] [CrossRef]
- Liang, R.; Li, X.; Li, W.; Zhu, X.; Li, C. DNA methylation in lung cancer patients: Opening a “window of life” under precision medicine. Biomed. Pharmacother. 2021, 144, 112202. [Google Scholar] [CrossRef]
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Sill, M.; Plass, C.; Pfister, S.M.; Lipka, D.B. Molecular tumor classification using DNA methylome analysis. Hum. Mol. Genet. 2020, 29, R205–R213. [Google Scholar] [CrossRef]
- Bailleux, C.; Lacroix, L.; Barranger, E.; Delaloge, S. Using methylation signatures on cell-free DNA for early cancer detection: A new era in liquid biopsy? Ann. Oncol. 2020, 31, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Kim, G.; Lee, T.R.; Ahn, J.M.; Park, H.; Park, S.R.; Song, K.B.; Jun, E.; Oh, D.; Lee, J.W.; et al. Integrative modeling of tumor genomes and epigenomes for enhanced cancer diagnosis by cell-free DNA. Nat. Commun. 2023, 14, 2017. [Google Scholar] [CrossRef] [PubMed]
- Constantin, N.; Sina, A.A.I.; Korbie, D.; Trau, M. Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies. Epigenomes 2022, 6, 6. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zeng, T.; Pan, X.; Guo, W.; Gan, Z.; Zhang, Y.; Huang, T.; Cai, Y.D. Screening Dys-Methylation Genes and Rules for Cancer Diagnosis by Using the Pan-Cancer Study. IEEE Access 2020, 8, 489–501. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, L.; Qiu, J.; Xu, J.; Yang, X.; Zhang, Y.; Tian, X.; Guan, X.; Cen, X.; Zhao, Y. Tumor biomarkers for diagnosis, prognosis and targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Kuze, Y.; Kobayashi, S.; Nakashima, M.; Morishima, S.; Kawamata, T.; Makiyama, J.; Suzuki, K.; Seki, M.; Abe, K.; et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 2024, 627, 221–228. [Google Scholar] [CrossRef]
- Costa, P.M.d.S.; Sales, S.L.A.; Pinheiro, D.P.; Pontes, L.Q.; Maranhão, S.S.; Pessoa, C.d.O.; Furtado, G.P.; Furtado, C.L.M. Epigenetic reprogramming in cancer: From diagnosis to treatment. Front. Cell Dev. Biol. 2023, 11, 1116805. [Google Scholar] [CrossRef]
- Zhao, A.; Zhou, H.; Yang, J.; Li, M.; Niu, T. Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies. Signal Transduct. Target. Ther. 2023, 8, 71. [Google Scholar] [CrossRef]
- Ramazi, S.; Dadzadi, M.; Sahafnejad, Z.; Allahverdi, A. Epigenetic regulation in lung cancer. MedComm 2023, 4, e401. [Google Scholar] [CrossRef]
- Sadida, H.Q.; Abdulla, A.; Marzooqi, S.A.; Hashem, S.; Macha, M.A.; Akil, A.S.A.S.; Bhat, A.A. Epigenetic modifications: Key players in cancer heterogeneity and drug resistance. Transl. Oncol. 2024, 39, 101821. [Google Scholar] [CrossRef] [PubMed]
- Tivey, A.; Lee, R.J.; Clipson, A.; Hill, S.M.; Lorigan, P.; Rothwell, D.G.; Dive, C.; Mouliere, F. Mining nucleic acid “omics” to boost liquid biopsy in cancer. Cell Rep. Med. 2024, 5, 101736. [Google Scholar] [CrossRef]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, S.; Hübschmann, D.; Raimondi, F.; Horak, P.; Warsow, G.; Fröhlich, M.; Klink, B.; Gieldon, L.; Hutter, B.; Kleinheinz, K.; et al. Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat. Commun. 2019, 10, 1635. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, H.; Wang, R.; Chen, Y.; Ouyang, X.; Li, W.; Sun, Y.; Peng, A. Cancer epigenetics: From laboratory studies and clinical trials to precision medicine. Cell Death Discov. 2024, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.L.; Athar, M.; James E Ferguson, I.I.I.; Varambally, S. EZH2-targeted therapies in cancer: Hype or a reality. Cancer Res. 2020, 80, 5449. [Google Scholar] [CrossRef] [PubMed]
- Budagaga, Y.; Sabet, Z.; Zhang, Y.; Novotná, E.; Hanke, I.; Rozkoš, T.; Hofman, J. Tazemetostat synergistically combats multidrug resistance by the unique triple inhibition of ABCB1, ABCC1, and ABCG2 efflux transporters in vitro and ex vivo. Biochem. Pharmacol. 2023, 216, 115769. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Davern, M.; Reynolds, J.V.; Donlon, N.E.; Lysaght, J. The impact of histone deacetylase inhibitors on immune cells and implications for cancer therapy. Cancer Lett. 2023, 559, 216121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xiao, Y.; Ding, Z.; Zhou, Y.; Fei, X.; Liu, J.; Su, Z.; Peng, X.; Chen, J. HDAC-targeting epigenetic modulators for cancer immunotherapy. Eur. J. Med. Chem. 2024, 265, 116129. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Wang, F.; Elhassan, R.M.; Cheng, Y.; Tang, X.; Chen, W.; Fang, H.; Hou, X. Targeting histone deacetylases for cancer therapy: Trends and challenges. Acta Pharm. Sin. B 2023, 13, 2425–2463. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef]
- Concannon, K.; Morris, B.B.; Gay, C.M.; Byers, L.A. Combining targeted DNA repair inhibition and immune-oncology approaches for enhanced tumor control. Mol. Cell 2023, 83, 660–680. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Qiao, X.; Fang, Y.; Guo, R.; Bai, P.; Liu, S.; Li, T.; Jiang, Y.; Wei, S.; Na, Z.; et al. Epigenetics-targeted drugs: Current paradigms and future challenges. Signal Transduct. Target. Ther. 2024, 9, 332. [Google Scholar] [CrossRef]
- Guo, R.; Li, J.; Hu, J.; Fu, Q.; Yan, Y.; Xu, S.; Wang, X.; Jiao, F. Combination of epidrugs with immune checkpoint inhibitors in cancer immunotherapy: From theory to therapy. Int. Immunopharmacol. 2023, 120, 110417. [Google Scholar] [CrossRef]
- Petronek, M.S.; Bayanbold, K.; Amegble, K.; Tomanek-Chalkley, A.M.; Allen, B.G.; Spitz, D.R.; Bailey, C.K. Evaluating the iron chelator function of sirtinol in non-small cell lung cancer. Front. Oncol. 2023, 13, 1185715. [Google Scholar] [CrossRef] [PubMed]
- Akbaribazm, M.; Khazaei, M.R.; Khazaei, F.; Khazaei, M. Doxorubicin and Trifolium pratense L. (Red clover) extract synergistically inhibits brain and lung metastases in 4T1 tumor-bearing BALB/c mice. Food Sci. Nutr. 2020, 8, 5557. [Google Scholar] [CrossRef]
- Li, M.; Hao, B.; Zhang, M.; Reiter, R.J.; Lin, S.; Zheng, T.; Chen, X.; Ren, Y.; Yue, L.; Abay, B.; et al. Melatonin enhances radiofrequency-induced NK antitumor immunity, causing cancer metabolism reprogramming and inhibition of multiple pulmonary tumor development. Signal Transduct. Target. Ther. 2021, 6, 330. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Sadida, H.Q.; Kuttikrishnan, S.; Junejo, K.; Bhat, A.A.; Uddin, S. Beyond genetics: Exploring the role of epigenetic alterations in breast cancer. Pathol.-Res. Pract. 2024, 254, 155174. [Google Scholar] [CrossRef]
- Babar, Q.; Saeed, A.; Tabish, T.A.; Pricl, S.; Townley, H.; Thorat, N. Novel epigenetic therapeutic strategies and targets in cancer. Biochim. Biophys. Acta-(BBA)-Mol. Basis Dis. 2022, 1868, 166552. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Agarwal, A.; Kansal, V.; Farooqi, H.; Prasad, R.; Singh, V.K. Epigallocatechin Gallate (EGCG), an Active Phenolic Compound of Green Tea, Inhibits Tumor Growth of Head and Neck Cancer Cells by Targeting DNA Hypermethylation. Biomedicines 2023, 11, 789. [Google Scholar] [CrossRef]
- Bontempo, P.; Capasso, L.; De Masi, L.; Nebbioso, A.; Rigano, D. Therapeutic Potential of Natural Compounds Acting through Epigenetic Mechanisms in Cardiovascular Diseases: Current Findings and Future Directions. Nutrients 2024, 16, 2399. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Bai, M.; Sun, Z.; Yao, N.; Zhang, A.; Guo, S.; Asemi, Z. Epigallocatechin-3-gallate and cancer: Focus on the role of microRNAs. Cancer Cell Int. 2023, 23, 241. [Google Scholar] [CrossRef] [PubMed]
- Volpes, S.; Cruciata, I.; Ceraulo, F.; Schimmenti, C.; Naselli, F.; Pinna, C.; Mauro, M.; Picone, P.; Dallavalle, S.; Nuzzo, D.; et al. Nutritional epigenomic and DNA-damage modulation effect of natural stilbenoids. Sci. Rep. 2023, 13, 658. [Google Scholar] [CrossRef] [PubMed]
- Ocaña-Paredes, B.; Rivera-Orellana, S.; Ramírez-Sánchez, D.; Montalvo-Guerrero, J.; Freire, M.P.; Espinoza-Ferrao, S.; Altamirano-Colina, A.; Echeverría-Espinoza, P.; Ramos-Medina, M.J.; Echeverría-Garcés, G.; et al. The pharmacoepigenetic paradigm in cancer treatment. Front. Pharmacol. 2024, 15, 1381168. [Google Scholar] [CrossRef]
- Bunnik, E.M.; Bolt, I.L. Exploring the Ethics of Implementation of Epigenomics Technologies in Cancer Screening: A Focus Group Study. Epigenetics Insights 2021, 14, 25168657211063618. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.N.G.; Kamphorst, B.A.; Mulder, B.C.; Kamp, I.v.; Boekhold, S.; Hazel, P.v.d.; Verweij, M.F. Ethics of early detection of disease risk factors: A scoping review. BMC Med. Ethics 2024, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Santaló, J.; Berdasco, M. Ethical implications of epigenetics in the era of personalized medicine. Clinical Epigenetics 2022, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Athieniti, E.; Spyrou, G.M. A guide to multi-omics data collection and integration for translational medicine. Comput. Struct. Biotechnol. J. 2022, 21, 134. [Google Scholar] [CrossRef]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.S.M.; Chang, H.Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Heining, C.; Kreutzfeldt, S.; Hutter, B.; Mock, A.; Hüllein, J.; Fröhlich, M.; Uhrig, S.; Jahn, A.; Rump, A.; et al. Comprehensive Genomic and Transcriptomic Analysis for Guiding Therapeutic Decisions in Patients with Rare Cancers. Cancer Discov. 2021, 11, 2780–2795. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, G.; Wang, J.; Zain, A.M.; Guo, W. Lung cancer subtype diagnosis using weakly-paired multi-omics data. Bioinformatics 2022, 38, 5092–5099. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef]
- Cazaly, E.; Saad, J.; Wang, W.; Heckman, C.; Ollikainen, M.; Tang, J. Making Sense of the Epigenome Using Data Integration Approaches. Front. Pharmacol. 2019, 10, 126. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| DNA Methylation Markers | RASSF1A (Ras Association Domain Family Member 1):
|
| Non-Coding RNA Markers | MicroRNAs (miRNAs):
|
| Histone Marks | H3K27me3 (Trimethylation of Histone H3 Lysine 27):
|
| Liquid Biopsy- Based Markers | Circulating Tumor DNA (ctDNA): Epigenetic markers in ctDNA, exosomes, or sputum are minimally invasive options [84]. Gene Panels: RASSF1A, SHOX2, and p16 methylation validated for lung cancer screening [85]. Methylation Signatures: Sputum methylation profiles help identify early-stage lung cancer in high-risk populations [86]. Diagnostic Tools: |
| Study Characteristics | Mechanism of Action | Therapeutic Focus |
|---|---|---|
| NCT03220477 https://clinicaltrials.gov/study/NCT03220477 Mocetinostat Phase I | Mocetinostat will be given as treatment and side effect observation | Pembrolizumab in Combination with Guadecitabine and Mocetinostat for patients with Advanced Lung Cancer |
| NCT05573035 https://clinicaltrials.gov/study/NCT05573035 LYL845 Phase I | Epigenetically reprogrammed tumor infiltrating lymphocyte therapy | Evaluate the safety and anti-tumor activity of LYL845 in participants with relapsed or refractory metastatic or locally advanced NSCLC |
| NCT06694454 https://clinicaltrials.gov/study/NCT06694454 AZA-AEGEAN Phase I/II | Inhaled Azacytidine With Platinum-Based Chemotherapy and Durvalumab | Determine the frequency of pathologic complete responses in participants for early-stage NSCLC |
| NCT02664181 https://clinicaltrials.gov/study/NCT02664181 Tetra-hydrouridine- decitabine (THU-Dec) Phase II | Investigation THU-Dec in combination with Nivolumab | Epigenetic immunotherapy for second line therapy in patients with NSCLC |
| NCT02546986 https://clinicaltrials.gov/study/NCT02546986 CC-486 Pembrolizumab Phase II | Assess the safety and efficacy of combination therapy | Epigenetic modulation and immune checkpoint therapy |
| NCT04814407 https://clinicaltrials.gov/study/NCT04814407 ctDNA Observational | Circulating Epigenetic Biomarkers | Identification of novel circulating methylated biomarkers for early lung cancer detection |
| NCT05707585 https://clinicaltrials.gov/study/NCT05707585 Biopsy Observational | Epigenetic Imprinting Biomarkers | Distinguish benign and malignant pulmonary nodules presurgically |
| NCT02259218 https://clinicaltrials.gov/study/NCT02259218 Molecular profiling Observational | Identification of predictive biomarkers for radiation toxicity and survival | Collected blood, urine, and tissue samples are analyzed for biomarkers via metabolomic and epigenetic profiling |
|
NCT06717243 https://clinicaltrials.gov/study/NCT06717243 Molecular profiling Observational | Genomic and Epigenetic Markers Associated with Resistance to Chemo-Immuno-therapy | Capture the dynamic changes that occur in the tumor microenvironment and how these relate to treatment outcomes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumann, A.A.; Buribayev, Z.; Wolkenhauer, O.; Salybekov, A.A.; Wolfien, M. Epigenomic Echoes—Decoding Genomic and Epigenetic Instability to Distinguish Lung Cancer Types and Predict Relapse. Epigenomes 2025, 9, 5. https://doi.org/10.3390/epigenomes9010005
Baumann AA, Buribayev Z, Wolkenhauer O, Salybekov AA, Wolfien M. Epigenomic Echoes—Decoding Genomic and Epigenetic Instability to Distinguish Lung Cancer Types and Predict Relapse. Epigenomes. 2025; 9(1):5. https://doi.org/10.3390/epigenomes9010005
Chicago/Turabian StyleBaumann, Alexandra A., Zholdas Buribayev, Olaf Wolkenhauer, Amankeldi A. Salybekov, and Markus Wolfien. 2025. "Epigenomic Echoes—Decoding Genomic and Epigenetic Instability to Distinguish Lung Cancer Types and Predict Relapse" Epigenomes 9, no. 1: 5. https://doi.org/10.3390/epigenomes9010005
APA StyleBaumann, A. A., Buribayev, Z., Wolkenhauer, O., Salybekov, A. A., & Wolfien, M. (2025). Epigenomic Echoes—Decoding Genomic and Epigenetic Instability to Distinguish Lung Cancer Types and Predict Relapse. Epigenomes, 9(1), 5. https://doi.org/10.3390/epigenomes9010005