
Experimental and Computational Approaches for Non-CpG Methylation Analysis


{kind=link}
{kind=link}
Abstract
1. Introduction
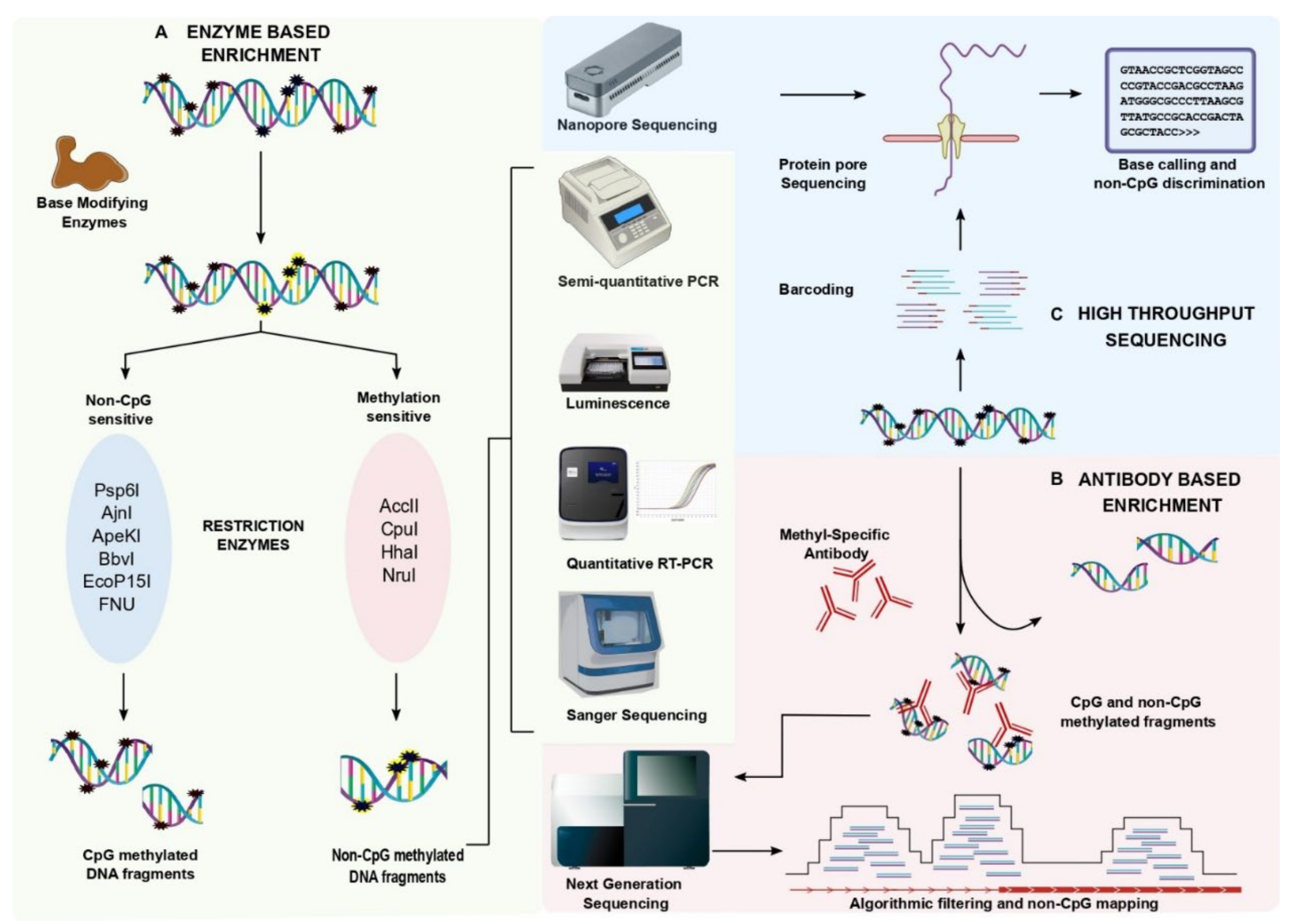
2. Experimental Approaches for Non-CpG Methylation Analysis
2.1. Conventional Methods
2.2. Enzyme-Based Enrichment Methods
2.3. Coupling Enzyme-Based Enrichment Strategies with High Throughput Sequencing
2.4. Antibody-Based Enrichment Methods
2.5. The New Era of Non-CpG Methylation Detection
3. Computational Approaches for Non-CpG Methylation
4. Current Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-mC | 5-methylcytosine |
| DNMT | DNA methyltransferases |
| PCR | Polymerase chain reaction |
| MBD | Methyl CpG binding domain captures protein |
| MeDIP | Methylated DNA immunoprecipitation |
| LUMA | Luminometric methylation assay |
| BS-seq | Bisulfite sequencing |
| RE | Restriction enzyme |
| WGBS | Whole-genome bisulfite sequencing |
| RRBS | Reduced representative bisulfite sequencing |
| SMRT | Single-molecule real-time sequencing |
References
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The Role of DNA Methylation in Mammalian Epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Müller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic Distribution and Inter-Sample Variation of Non-CpG Methylation across Human Cell Types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, D.; Deva Magendhra Rao, A.K.; Rajkumar, T.; Mani, S. Non-CpG Methylation—A Key Epigenetic Modification in Cancer. Brief. Funct. Genomics 2021, 20, 304–311. [Google Scholar] [CrossRef]
- Fuso, A. Non-CpG Methylation Revised. Epigenomes 2018, 2, 22. [Google Scholar] [CrossRef]
- Imamura, T.; Kerjean, A.; Heams, T.; Kupiec, J.J.; Thenevin, C.; Pàldi, A. Dynamic CpG and Non-CpG Methylation of the Peg1/Mest Gene in the Mouse Oocyte and Preimplantation Embryo. J. Biol. Chem. 2005, 280, 20171–20175. [Google Scholar] [CrossRef]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the Unique DNA Methylation Landscape of the Brain: Non-CpG Methylation, Hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef]
- Shirane, K.; Toh, H.; Kobayashi, H.; Miura, F.; Chiba, H.; Ito, T.; Kono, T.; Sasaki, H. Mouse Oocyte Methylomes at Base Resolution Reveal Genome-Wide Accumulation of Non-CpG Methylation and Role of DNA Methyltransferases. PLoS Genet. 2013, 9, e1003439. [Google Scholar] [CrossRef]
- Ichiyanagi, T.; Ichiyanagi, K.; Miyake, M.; Sasaki, H. Accumulation and Loss of Asymmetric Non-CpG Methylation during Male Germ-Cell Development. Nucleic Acids Res. 2013, 41, 738–745. [Google Scholar] [CrossRef]
- Kouidou, S.; Agidou, T.; Kyrkou, A.; Andreou, A.; Katopodi, T.; Georgiou, E.; Krikelis, D.; Dimitriadou, A.; Spanos, P.; Tsilikas, C.; et al. Non-CpG Cytosine Methylation of P53 Exon 5 in Non-Small Cell Lung Carcinoma. Lung Cancer 2005, 50, 299–307. [Google Scholar] [CrossRef]
- Li, C.; Xiong, W.; Liu, X.; Xiao, W.; Guo, Y.; Tan, J.; Li, Y. Hypomethylation at Non-CpG/CpG Sites in the Promoter of HIF-1α Gene Combined with Enhanced H3K9Ac Modification Contribute to Maintain Higher HIF-1α Expression in Breast Cancer. Oncogenesis 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Lucarelli, M. CpG and Non-CpG Methylation in the Diet-Epigenetics-Neurodegeneration Connection. Curr. Nutr. Rep. 2019, 8, 74–82. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.J.; Nakai, K. Differential Landscape of Non-CpG Methylation in Embryonic Stem Cells and Neurons Caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Stein, R.; Cedar, H.; Razin, A. Methylation of CpG Sequences in Eukaryotic DNA. FEBS Lett. 1981, 124, 67–71. [Google Scholar] [CrossRef]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG Methylation Is Prevalent in Embryonic Stem Cells and May Be Mediated by DNA Methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef]
- Malone, C.S.; Miner, M.D.; Doerr, J.R.; Jackson, J.P.; Jacobsen, S.E.; Wall, R.; Teitell, M. CmC(A/T)GG DNA Methylation in Mature B Cell Lymphoma Gene Silencing. Proc. Natl. Acad. Sci. USA 2001, 98, 10404–10409. [Google Scholar] [CrossRef]
- Rein, T.; DePamphilis, M.L.; Zorbas, H. Identifying 5-Methylcytosine and Related Modifications in DNA Genomes. Nucleic Acids Res. 1998, 26, 2255–2264. [Google Scholar] [CrossRef]
- Lyko, F.; Ramsahoye, B.H.; Jaenisch, R. DNA Methylation in Drosophila Melanogaster. Nature 2000, 408, 538–540. [Google Scholar] [CrossRef]
- Castel, A.L.; Nakamori, M.; Thornton, C.A.; Pearson, C.E. Identification of Restriction Endonucleases Sensitive to 5-Cytosine Methylation at Non-CpG Sites, Including Expanded (CAG)n/(CTG)n Repeats. Epigenetics 2011, 6, 416. [Google Scholar] [CrossRef]
- Melnikov, A.A.; Gartenhaus, R.B.; Levenson, A.S.; Motchoulskaia, N.A.; Levenson, V.V. MSRE-PCR for Analysis of Gene-Specific DNA Methylation. Nucleic Acids Res. 2005, 33, e93. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Johansson, S.; Ekström, T.J. Using LUMA: A Luminometric-Based Assay for Global DNA-Methylation. Epigenetics 2006, 1, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Gu, H.; Bock, C.; Gnirke, A.; Meissner, A. High-Throughput Bisulfite Sequencing in Mammalian Genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef] [PubMed]
- How-Kit, A.; Daunay, A.; Mazaleyrat, N.; Busato, F.; Daviaud, C.; Teyssier, E.; Deleuze, J.F.; Gallusci, P.; Tost, J. Accurate CpG and Non-CpG Cytosine Methylation Analysis by High-Throughput Locus-Specific Pyrosequencing in Plants. Plant Mol. Biol. 2015, 88, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Ferraguti, G.; Scarpa, S.; Ferrer, I.; Lucarelli, M. Disclosing Bias in Bisulfite Assay: MethPrimers Underestimate High DNA Methylation. PLoS ONE 2015, 10, e0118318. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-Scale DNA Methylation Maps of Pluripotent and Differentiated Cells. Nature 2008, 454, 766. [Google Scholar] [CrossRef]
- Patalano, S.; Vlasova, A.; Wyatt, C.; Ewels, P.; Camara, F.; Ferreira, P.G.; Asher, C.L.; Jurkowski, T.P.; Segonds-Pichon, A.; Bachman, M.; et al. Molecular Signatures of Plastic Phenotypes in Two Eusocial Insect Species with Simple Societies. Proc. Natl. Acad. Sci. USA 2015, 112, 13970–13975. [Google Scholar] [CrossRef]
- Ross, S.E.; Angeloni, A.; Geng, F.S.; de Mendoza, A.; Bogdanovic, O. Developmental Remodelling of Non-CG Methylation at Satellite DNA Repeats. Nucleic Acids Res. 2020, 48, 12675–12688. [Google Scholar] [CrossRef]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of Whole-Genome Bisulfite Sequencing Library Preparation Strategies Identifies Sources of Biases Affecting DNA Methylation Data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef]
- Laird, C.D.; Pleasant, N.D.; Clark, A.D.; Sneeden, J.L.; Hassan, K.M.A.; Manley, N.C.; Vary, J.C.; Morgan, T.; Hansen, R.S.; Stöger, R. Hairpin-Bisulfite PCR: Assessing Epigenetic Methylation Patterns on Complementary Strands of Individual DNA Molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 204–209. [Google Scholar] [CrossRef]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In Vivo Control of CpG and Non-CpG DNA Methylation by DNA Methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.E.; Hesselson, D.; Bogdanovic, O. Developmental Accumulation of Gene Body and Transposon Non-CpG Methylation in the Zebrafish Brain. Front. Cell Dev. Biol. 2021, 9, 643603. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Ward, R.L.; Hesson, L.B. The Evidence for Functional Non-CpG Methylation in Mammalian Cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, Recognition and Regulation of Non-CpG Methylation in the Adult Mammalian Brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, B.; Xing, X.; Wang, T. Combining MeDIP-Seq and MRE-Seq to Investigate Genome-Wide CpG Methylation. Methods 2015, 72, 29–40. [Google Scholar] [CrossRef]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of Methyl-DNA Immunoprecipitation (MeDIP) and Methyl-CpG Binding Domain (MBD) Protein Capture for Genome-Wide DNA Methylation Analysis Reveal CpG Sequence Coverage Bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Nagarajan, R.P.; Costello, J.F. Genome-Scale DNA Methylation Analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef]
- Robertson, J.; Robertson, A.B.; Klungland, A. The Presence of 5-Hydroxymethylcytosine at the Gene Promoter and Not in the Gene Body Negatively Regulates Gene Expression. Biochem. Biophys. Res. Commun. 2011, 411, 40–43. [Google Scholar] [CrossRef][Green Version]
- Goldsmith, C.; Rodríguez-Aguilera, J.R.; El-Rifai, I.; Jarretier-Yuste, A.; Hervieu, V.; Raineteau, O.; Saintigny, P.; Chagoya de Sánchez, V.; Dante, R.; Ichim, G.; et al. Low Biological Fluctuation of Mitochondrial CpG and Non-CpG Methylation at the Single-Molecule Level. Sci. Rep. 2021, 11, 8032. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Kishore, K.; de Pretis, S.; Lister, R.; Morelli, M.J.; Bianchi, V.; Amati, B.; Ecker, J.R.; Pelizzola, M. MethylPipe and CompEpiTools: A Suite of R Packages for the Integrative Analysis of Epigenomics Data. BMC Bioinform. 2015, 16, 313. [Google Scholar] [CrossRef] [PubMed]
- Catoni, M.; Tsang, J.M.F.; Greco, A.P.; Zabet, N.R. DMRcaller: A Versatile R/Bioconductor Package for Detection and Visualization of Differentially Methylated Regions in CpG and Non-CpG Contexts. Nucleic Acids Res. 2018, 46, e114. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.S.; Wu, B.H.; Yen, M.R.; Chen, P.Y. MethGET: Web-Based Bioinformatics Software for Correlating Genome-Wide DNA Methylation and Gene Expression. BMC Genom. 2020, 21, 375. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramasamy, D.; Rao, A.K.D.M.; Rajkumar, T.; Mani, S. Experimental and Computational Approaches for Non-CpG Methylation Analysis. Epigenomes 2022, 6, 24. https://doi.org/10.3390/epigenomes6030024
Ramasamy D, Rao AKDM, Rajkumar T, Mani S. Experimental and Computational Approaches for Non-CpG Methylation Analysis. Epigenomes. 2022; 6(3):24. https://doi.org/10.3390/epigenomes6030024
Chicago/Turabian StyleRamasamy, Deepa, Arunagiri Kuha Deva Magendhra Rao, Thangarajan Rajkumar, and Samson Mani. 2022. "Experimental and Computational Approaches for Non-CpG Methylation Analysis" Epigenomes 6, no. 3: 24. https://doi.org/10.3390/epigenomes6030024
APA StyleRamasamy, D., Rao, A. K. D. M., Rajkumar, T., & Mani, S. (2022). Experimental and Computational Approaches for Non-CpG Methylation Analysis. Epigenomes, 6(3), 24. https://doi.org/10.3390/epigenomes6030024