
Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies



Abstract
:1. Introduction
2. Challenges Associated with the Current Screening Paradigm to Efficiently Identify Early-Stage Malignancies
3. The Clinical Potential of Implementing a Single Test for Multiple Cancer Early Detection (stMCED)
4. The Potential Value of Methylated-cfDNA for Developing stMCED
5. Criteria for Developing Efficient stMCED
6. Methodologies for stMCED Screening
6.1. Non-Methylation Based Assays
6.1.1. DEEPGEN™
6.1.2. CancerSEEK
6.2. Methylation-Based Assay
6.2.1. PanSEER
6.2.2. cfMeDIP-Seq
6.2.3. IvyGene®
6.2.4. GRAIL
6.2.5. Methylscape
7. Clinical Translation of stMCEDs: Summary and Future Perspectives
8. Methodology
8.1. Literature Search
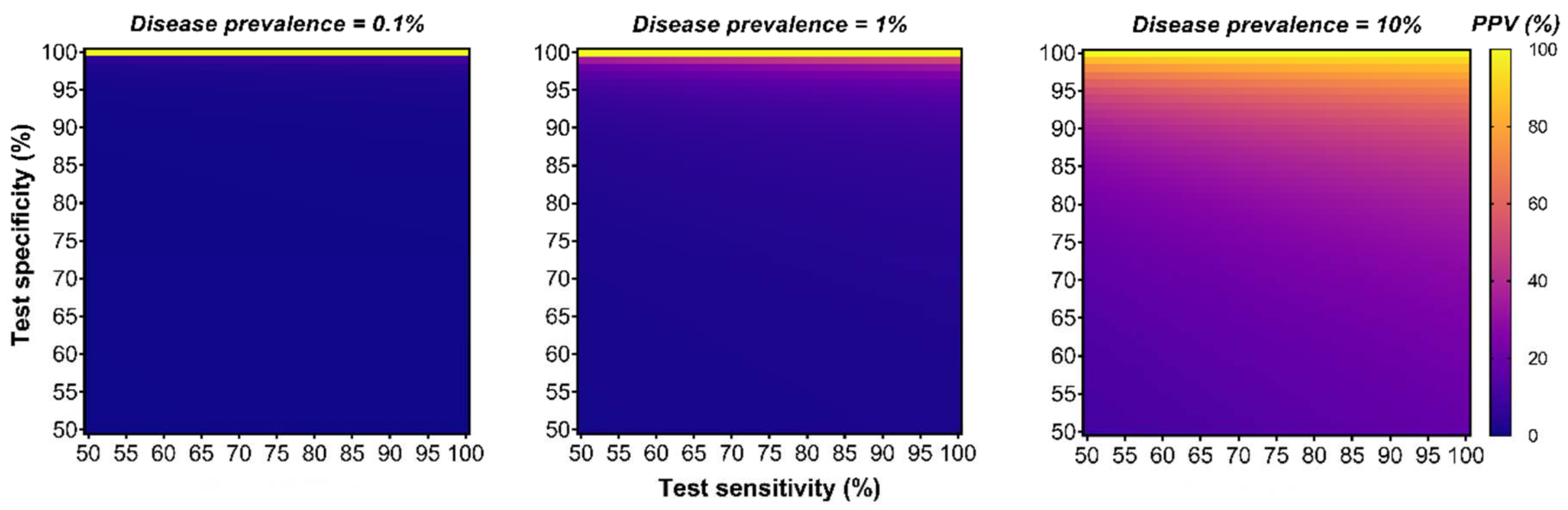
8.2. Positive Predictive Value (PPV) Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, A.B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, 1–22. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guide to Cancer-Guide to Cancer Early Diagnosis; WHO: Geneva, Switzerland, 2017; ISBN 9789241511940. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, R.; Urban, N.; Ramsey, S.; McIntosh, M.; Schwartz, S.; Reid, B.; Radich, J.; Anderson, G.; Hartwell, L. The case for early detection. Nat. Rev. Cancer 2003, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, A.; Cohen, S.S.; Reichert, H.; Kansal, A.R.; Chung, K.C.; Ofman, J.J. Estimating the population health impact of a multi-cancer early detection genomic blood test to complement existing screening in the US and UK. Br. J. Cancer 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Perakis, S.; Geigl, J.B.; Speicher, M.R. The potential of liquid biopsies for the early detection of cancer. NPJ Precis. Oncol. 2017, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Marrugo-Ramírez, J.; Mir, M.; Samitier, J. Blood-based cancer biomarkers in liquid biopsy: A promising non-invasive alternative to tissue biopsy. Int. J. Mol. Sci. 2018, 19, 2877. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Speicher, M.R. The biology of circulating tumor cells. Oncogene 2016, 35, 1216–1224. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Brock, G.; Castellanos-Rizaldos, E.; Hu, L.; Coticchia, C.; Skog, J. Liquid biopsy for cancer screening, patient stratification and monitoring. Transl. Cancer Res. 2015, 4, 280–290. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, D. Emerging molecular markers of cancer. Nat. Rev. Cancer 2002, 2, 210–219. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Park, Y.J.; Plass, C. Epigenetic reprogramming in cancer. Epigenet. Hum. Health 2015, 4, 193–223. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Roy, D. Maarit Tiirikainen Diagnostic Power of DNA Methylation Classifiers for Early Detection of Cancer. Trends Cancer 2020, 78–81. [Google Scholar] [CrossRef]
- Smith, R.A.; Andrews, K.S.; Brooks, D.; Fedewa, S.A.; Manassaram-Baptiste, D.; Saslow, D.; Wender, R.C. Cancer screening in the United States, 2019: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2019, 69, 184–210. [Google Scholar] [CrossRef]
- Lehman, C.D.; Arao, R.F.; Sprague, B.L.; Lee, J.M.; Buist, D.S.M.; Kerlikowske, K.; Henderson, L.M.; Tosteson, A.N.A.; Rauscher, G.H.; Miglioretti, D.L. National Performance Benchmarks for Modern Screening Digital Mammography. Radiology 2017, 283, 49–58. [Google Scholar] [CrossRef]
- Wolf., A.M.; Wender, R.C.; Etzioni, R.B.; Thompson, I.M.; D’Amico, A.V.; Volk, R.J.; Brooks, D.D.; Dash, C.; Guessous, I.; Andrews, K.; et al. American Cancer Society guideline for the early detection of prostate cancer: Update 2010. CA Cancer J. Clin. 2010, 47, 70–98. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.M.D.; Fontham, E.T.H.; Church, T.R.; Flowers, C.R.; Guerra, C.E.; LaMonte, S.J.; Etzioni, R.; McKenna, M.T.; Oeffinger, K.C.; Shih, Y.-C.T.; et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018, 68, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.S.; Piper, M.A.; Perdue, L.A.; Rutter, C.; Webber, E.M.; O’Connor, E.; Smith, N.; Whitlock, E.P. Screening for Colorectal Cancer: A Systematic Review for the U.S. Preventive Services Task Force. In Evidence Syntheses, No. 135; AHRQ Publ. No. 14-05203-EF-1; Agency Healthcare Research and Quality: Rockville, MD, USA, 2016; p. 239. [Google Scholar]
- Curry, S.J.; Krist, A.H.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W.; Kemper, A.R.; Kubik, M.; et al. Screening for cervical cancer us preventive services task force recommendation statement. JAMA J. Am. Med. Assoc. 2018, 320, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Koliopoulos, G.; Arbyn, M.; Martin-Hirsch, P.; Kyrgiou, M.; Prendiville, W.; Paraskevaidis, E. Diagnostic accuracy of human papillomavirus testing in primary cervical screening: A systematic review and meta-analysis of non-randomized studies. Gynecol. Oncol. 2007, 104, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, P.F.; Gierada, D.S.; Black, W.; Munden, R.; Nath, H.; Aberle, D.; Kazerooni, E. Performance of lung-RADS in the national lung screening trial: A retrospective assessment. Ann. Intern. Med. 2015, 162, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croswell, J.M.; Kramer, B.S.; Kreimer, A.R.; Prorok, P.C.; Xu, J.L.; Baker, S.G.; Fagerstrom, R.; Riley, T.L.; Clapp, J.D.; Berg, C.D.; et al. Cumulative incidence of false-positive results in repeated, multimodal cancer screening. Ann. Fam. Med. 2009, 7, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.A.; Maple, J.T.; Ben-Menachem, T.; Cash, B.D.; Decker, G.A.; Early, D.S.; Evans, J.A.; Fanelli, R.D.; Fukami, N.; Hwang, J.H.; et al. Complications of colonoscopy. Gastrointest. Endosc. 2011, 74, 745–752. [Google Scholar] [CrossRef]
- Korfage, I.J.; Van Ballegooijen, M.; Wauben, B.; Looman, C.W.N.; Habbema, J.D.F.; Essink-Bot, M.L. Having a Pap smear, quality of life before and after cervical screening: A questionnaire study. BJOG An. Int. J. Obstet. Gynaecol. 2012, 119, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.J. Radiation Risks Potentially Associated with Low-Dose CT Screening of Adult Smokers for Lung Cancer. Radiology 2004, 231, 440–445. [Google Scholar] [CrossRef]
- Ali, R.M.K.M.; England, A.; McEntee, M.F.; Mercer, C.E.; Tootell, A.; Hogg, P. Effective lifetime radiation risk for a number of national mammography screening programmes. Radiography 2018, 24, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Glas, A.S.; Lijmer, J.G.; Prins, M.H.; Bonsel, G.J.; Bossuyt, P.M.M. The diagnostic odds ratio: A single indicator of test performance. J. Clin. Epidemiol. 2003, 56, 1129–1135. [Google Scholar] [CrossRef]
- Tenny, S.; Hoffman, M.R. Prevalence; StatPearls Publishing: Treasure Island, FL, USA, 2017. [Google Scholar]
- Clarke, C.A.; Hubbell, E.; Kurian, A.W.; Colditz, G.A.; Hartman, A.-R.; Gomez, S.L. Projected Reductions in Absolute CancerRelated Deaths from Diagnosing Cancers Before Metastasis, 2006–2015. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, E.; Clarke, C.A.; Aravanis, A.M.; Berg, C.D. Modeled reductions in late-stage cancer with a multi-cancer early detection test. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Gulati, R.; Katki, H.A.; Castle, P.E.; Etzioni, R. A Quantitative Framework to Study Potential Benefits and Harms of Multi-cancer Early Detection Testing. Cancer Epidemiol. Biomarkers Prev. 2022, 31, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hesson, L.B.; Pritchard, A.L. Clinical Epigenetics; Springer Nature: Berlin/Heidelberg, Germany, 2019; ISBN 9789811389573. [Google Scholar]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.S. DNA methylation age of human tissues and cell type. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Taryma-Leśniak, O.; Sokolowska, K.E.; Wojdacz, T.K. Current status of development of methylation biomarkers for in vitro diagnostic IVD applications. Clin. Epigenet. 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Constâncio, V.; Nunes, S.P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9, 624. [Google Scholar] [CrossRef] [Green Version]
- Feber, A.; Dhami, P.; Dong, L.; de Winter, P.; Tan, W.S.; Martínez-Fernández, M.; Paul, D.S.; Hynes-Allen, A.; Rezaee, S.; Gurung, P.; et al. UroMark—A urinary biomarker assay for the detection of bladder cancer. Clin. Epigenet. 2017, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. 2017, 12, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.D.; Oh, T.J.; Chung, T.H.; Jang, H.W.; Kim, Y.N.; An, S.; Kim, N.K. Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin. Epigenet. 2019, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oussalah, A.; Rischer, S.; Bensenane, M.; Conroy, G.; Filhine-Tresarrieu, P.; Debard, R.; Forest-Tramoy, D.; Josse, T.; Reinicke, D.; Garcia, M.; et al. Plasma mSEPT9: A Novel Circulating Cell-free DNA-Based Epigenetic Biomarker to Diagnose Hepatocellular Carcinoma. EBioMedicine 2018, 30, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, G.D.; Ofman, J.J. Criteria for Evaluating Multi-cancer Early Detection Tests. Oncol. Haematol. 2021, 17, 3. [Google Scholar] [CrossRef]
- Ris, F.; Hellan, M.; Douissard, J.; Nieva, J.J.; Triponez, F.; Woo, Y.; Geller, D.; Buchs, N.C.; Buehler, L.; Moenig, S.; et al. Blood-Based Multi-Cancer Detection Using a Novel Variant Calling Assay (DEEPGENTM): Early Clinical Results. Cancers 2021, 13, 4104. [Google Scholar] [CrossRef]
- Hermann, B.T.; Pfeil, S.; Groenke, N.; Schaible, S.; Kunze, R.; Hagen, M.E.; Bhakdi, J. DEEPGENTM—A Novel Variant Calling Assay for Low Frequency Variants. Genes 2021, 12, 507. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Taggart, D.; Zheng, L.; Liu, D.; Li, G.; Li, M.; Zhang, K.; Van Etten, R.A. Circulating cell-free DNA methylation assay: Towards early detection of multiple cancer types. In Proceedings of the The American Association for Cancer Research Annual Meeting, AACR, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Liu, M.C.; Klein, E.; Hubbell, E.; Maddala, T.; Aravanis, A.M.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K.; et al. Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Ann. Oncol. 2018, 29, viii14. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Sina, A.A.I.; Carrascosa, L.G.; Liang, Z.; Grewal, Y.S.; Wardiana, A.; Shiddiky, M.J.A.; Gardiner, R.A.; Samaratunga, H.; Gandhi, M.K.; Scott, R.J.; et al. Epigenetically reprogrammed methylation landscape drives the DNA self-assembly and serves as a universal cancer biomarker. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sina, A.A.I.; Carrascosa, L.G.; Trau, M. DNA Methylation-Based Point-of-Care Cancer Detection: Challenges and Possibilities. Trends Mol. Med. 2019, 25, 955–966. [Google Scholar] [CrossRef]
- Cancer Australia’s National Cancer Control Indicators (NCCI). Available online: https://ncci.canceraustralia.gov.au/outcomes/prevalence/five-year-prevalence (accessed on 3 December 2021).
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 2018, 23, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
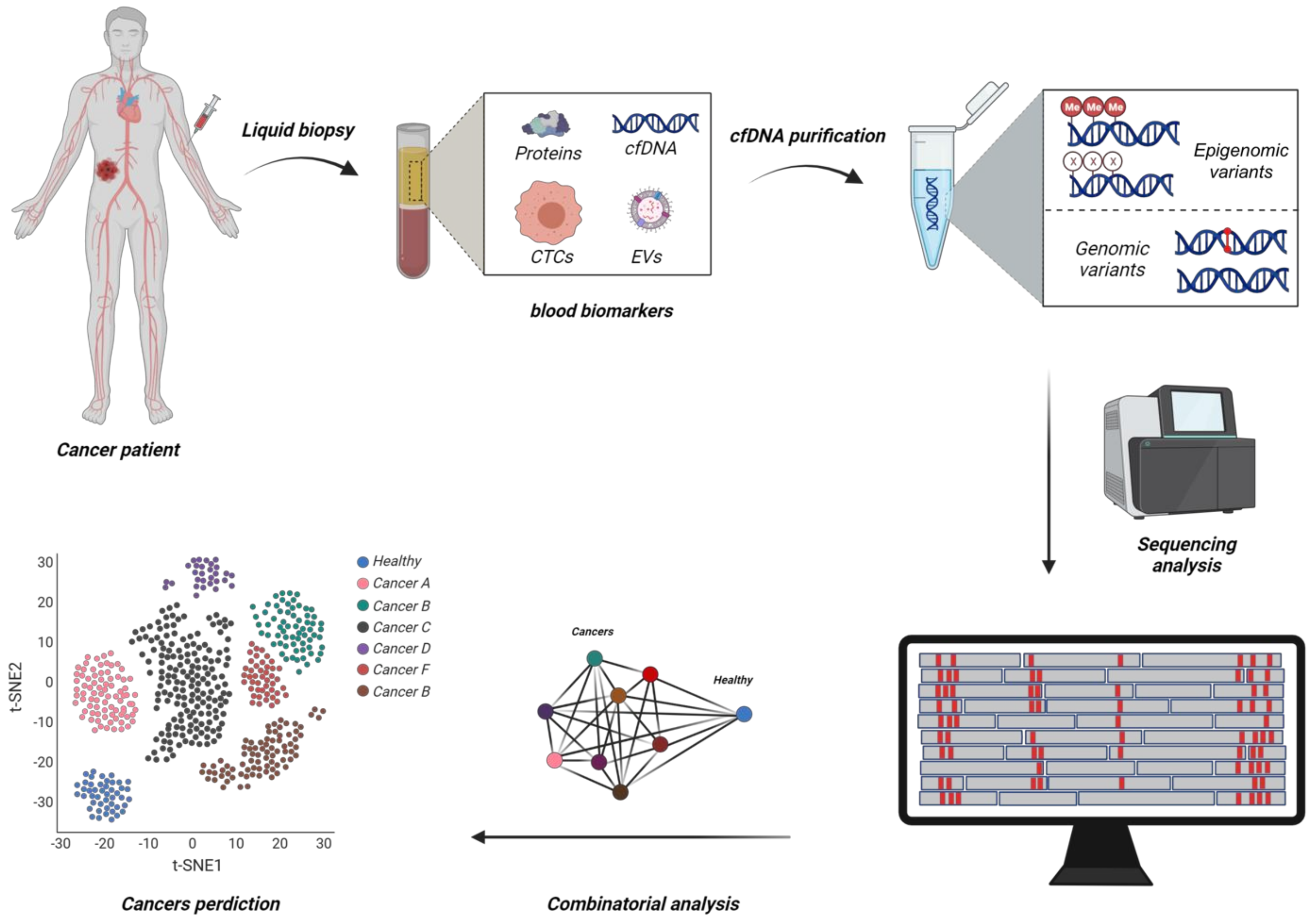


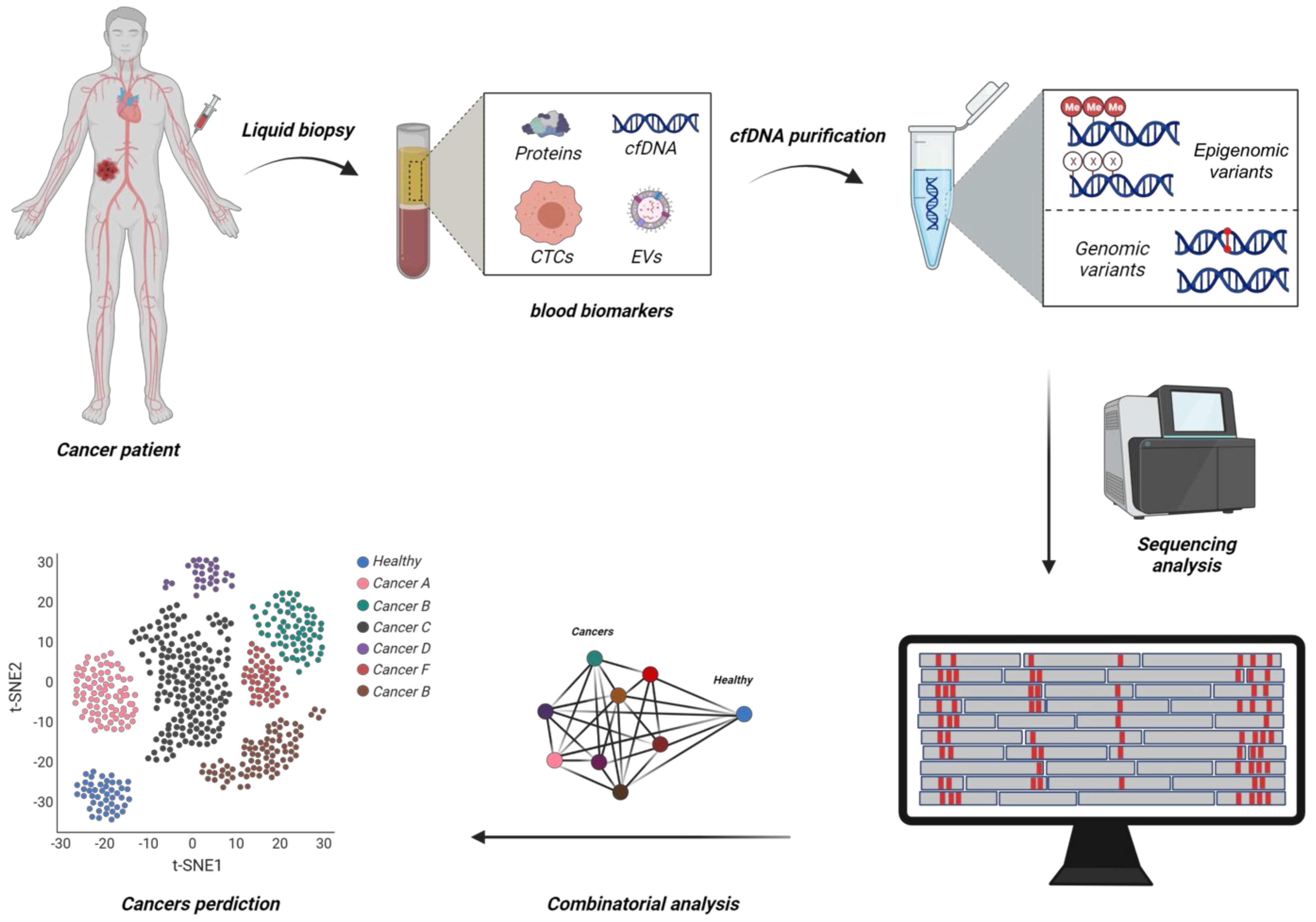{kind=link}
{kind=link}
| DEEPGEN™ | CancerSEEK | PanSEEER | cfMeDIP-seq | GRAIL | IvyGene® | |
|---|---|---|---|---|---|---|
| Biomarker type | Genomic variants | Genomic variant (1933 mutations, 16 genes) & 8 proteins | 477 DMRs (657 genes, 10,613 CpGs) | Enriched DMRs | >100,000 DMRs (1,166,720 CpGs, cover 17.2 Mb) | Targeted panels of methylation biomarkers |
| Targeted cancer types | 7 | 8 | 5 | 9 (across 3 sperate studies) | 12 pre-specified (>50 sub-types) | 3 |
| Specificity (%) | 95 | 99.14 | 96.1 | - | 99.52 | 96–100 |
| Sensitivity (%): overall stage 1 stage 2 stage 3 stage 4 | 57 51 58 62 67 | 62.3 48 63 70 - | ~95 ~95 (s1-2) ~95 (s3-4) | - - - - - | 51.5 16.8 40.4 77 90.1 | 89–95 - - - - |
| AUROC: overall stage 1 stage 2 stage 3 stage 4 | 0.9 0.88 0.9 0.92 0.94 | 0.91 - - - - | ~0.99 ~0.99 (s1-2) ~0.99 (s3-4) | 0.91 to 0.99 (s1-2) 0.92 to 0.99 (s3-4) | - | - |
| TOO capacity (depends on organs) | No | Yes (median 63%) | No | Only | Yes (overall 88.7%) | Only |
| cfDNA input (ng) | - | - | ~12 | 1–10 | - | - |
| LOD (% ctDNA) | > 0.09 | - | >0.01 | >0.001 | - | - |
| DEEPGEN™ | Cohort Size (Healthy = 415) | Specificity (%) | Sensitivity (%) | AUROC |
|---|---|---|---|---|
| All cancer | ||||
| stage 1 | 70 | 95 | 51 | 0.88 |
| stage 2 | 55 | 95 | 58 | 0.9 |
| stage 3 | 73 | 95 | 62 | 0.92 |
| stage 4 | 27 | 95 | 67 | 0.94 |
| Overall | 260 | 95/99 | 57/43 | 0.9 |
| Bladder | ||||
| Overall | 25 | 95/99 | 80/32 | |
| Prostate | ||||
| Overall | 29 | 95/99 | 72/62 | |
| Lung | ||||
| Overall | 30 | 95/99 | 67/53 | |
| Liver | ||||
| Overall | 27 | 95/99 | 63/41 | |
| Pancreatic | ||||
| Overall | 40 | 95/99 | 52/38 | |
| Colorectal | ||||
| Overall | 66 | 95/99 | 42/27 | |
| Breast | ||||
| Overall | 43 | 95/99 | 30/16 |
| CancerSEEK | Cohort Size (Healthy = 812) | Specificity (%) | Sensitivity (%) | AUROC | TOO Prediction |
|---|---|---|---|---|---|
| All cancer | 99.14 | ||||
| stage 1 | 199 | 48 | |||
| stage 2 | 497 | 63 | |||
| stage 3 | 309 | 70 | |||
| Overall | 1005 | 62.3 | 0.91 | 63% | |
| Ovary cancer | |||||
| stage 1 | 9 | 88.9 | |||
| stage 2 | 4 | 100.0 | |||
| stage 3 | 41 | 100.0 | |||
| Overall | 54 | 98.1 | 79% | ||
| Esophagus cancer | |||||
| stage 1 | 5 | 20.0 | |||
| stage 2 | 29 | 86.2 | |||
| stage 3 | 11 | 45.5 | |||
| Overall | 45 | 68.9 | 46% (with stomach) | ||
| Lung cancer | |||||
| stage 1 | 46 | 43.5 | |||
| stage 2 | 27 | 66.7 | |||
| stage 3 | 31 | 74.2 | |||
| Overall | 104 | 58.7 | 39% | ||
| Liver cancer | |||||
| stage 1 | 5 | 100.0 | |||
| stage 2 | 19 | 100.0 | |||
| stage 3 | 20 | 95.0 | |||
| Overall | 44 | 97.7 | 44% | ||
| Pancreatic cancer | |||||
| stage 1 | 4 | 25.0 | |||
| stage 2 | 83 | 73.5 | |||
| stage 3 | 6 | 83.3 | |||
| Overall | 93 | 72.0 | 81% | ||
| Colorectal cancer | |||||
| stage 1 | 77 | 42.9 | |||
| stage 2 | 191 | 72.3 | |||
| stage 3 | 120 | 67.5 | |||
| Overall | 388 | 64.9 | 84% | ||
| Breast cancer | |||||
| stage 1 | 32 | 37.5 | |||
| stage 2 | 114 | 25.4 | |||
| stage 3 | 63 | 46.0 | |||
| Overall | 209 | 33.5 | 63% | ||
| Stomach cancer | |||||
| stage 1 | 21 | 71.4 | |||
| stage 2 | 30 | 66.7 | |||
| stage 3 | 17 | 82.4 | |||
| Overall | 68 | 72.1 | 46% (with oesophagus) |
| PanSEER | Cohort Size (Healthy = 207) | Sample Number Per Stage: (1–2)–(3–4) | Specificity (%) | Sensitivity (%) | AUROC |
|---|---|---|---|---|---|
| All cancer | 96.10 | ||||
| Post diagnosis | 113 | 32–80 | 87.6 | 0.97 | |
| Pre diagnosis: | 98 | 94.9 | 0.99 | ||
| 0–1 year before | 21 | 5–13 | 95.2 | 0.99 | |
| 1–2 year before | 23 | 6–17 | 95.7 | 0.99 | |
| 2–3 years before | 31 | 10–17 | 93.6 | 0.99 | |
| 3–4 years before | 23 | 8–9 | 95.7 | 0.99 | |
| Esophagus | |||||
| stage 1–2 | 46 | ||||
| stage 3–4 | 63 | ||||
| Overall | 113 | ||||
| Lung | |||||
| stage 1–2 | 18 | ||||
| stage 3–4 | 80 | ||||
| Overall | 103 | ||||
| Liver | |||||
| stage 1–2 | 7 | ||||
| stage 3–4 | 43 | ||||
| Overall | 52 | ||||
| Colorectal | |||||
| stage 1–2 | 21 | ||||
| stage 3–4 | 16 | ||||
| Overall | 42 | ||||
| Stomach | |||||
| stage 1–2 | 44 | ||||
| stage 3–4 | 54 | ||||
| Overall | 104 |
| cfMeDIP-seq | Cohort Size in Sets: (Train/Test)—Validation | Accuracy to Predict Cancer with TOO (AUROC) |
|---|---|---|
| Lung cancer | ||
| stage 1–2 | 32 | 0.975 |
| stage 3–4 | (22)–23 | 0.966 |
| Overall | (25)–55 | 0.971 |
| Pancreatic cancer | ||
| stage 1–2 | (23)–15 | 0.914 |
| stage 3–4 | (1)–32 | 0.92 |
| Overall | (24)–47 | 0.918 |
| Acute myeloid leukaemia | ||
| Overall | 35 | 0.98 |
| Healthy | ||
| Overall | (24)–62 | 0.969 |
| Colorectal cancer | ||
| stage 1–2 | (1) | - |
| stage 3–4 | (21) | - |
| Overall | (23) | - |
| Bladder cancer | ||
| Overall | (20) | - |
| Renal cancer | ||
| Overall | (20) | - |
| Renal cancer | ||
| stage 1–2 | (33) | - |
| stage 3–4 | (66) | - |
| Overall | (99) | 0.99 |
| Intracranial Glioma | ||
| Overall | (59) | 0.99 |
| IvyGene® (Laboratory for Advanced Medicine) | Cohort Size | Specificity (%) | Sensitivity (%): Predict Cancer & TOO Accuracy |
|---|---|---|---|
| Liver cancer | |||
| Overall (stage 1–4) | 60 | 97.5 | 95 |
| Healthy (control) | |||
| Overall | 30 | ||
| Benign liver (control) | |||
| Overall | 10 | ||
| Other cancers (control) | |||
| Overall | 30 | ||
| Breast cancer | |||
| Overall (stage I-IV) | 65 | 96 | 89 |
| Healthy (control) | |||
| Overall | 39 | 95 | |
| Benign breast (control) | |||
| Overall | 15 | 100 | |
| Other cancers (control) | |||
| colorectal | 11 | ||
| liver | 9 | ||
| lung | 12 | ||
| Overall | 32 | 96 | |
| Colorectal cancer | |||
| Overall (stage 1–4) | 68 | 100 | 93 (67–100) |
| Healthy (control) | |||
| Overall | 42 | ||
| Benign colorectal (control) | |||
| Overall | 14 | ||
| Other cancers (control) | |||
| breast | 10 | 100 | |
| liver | 10 | 100 | |
| lung | 10 | 100 | |
| Overall | 30 |
| GRAIL | Cohort Size (Healthy = 1254) | Specificity (%) | Sensitivity (%) | TOO Prediction Accuracy (%) (For True Positive) |
|---|---|---|---|---|
| All cancer | 99.52 | |||
| Stage 1 | 849 | 16.8 | ||
| Stage 2 | 703 | 40.4 | ||
| Stage 3 | 566 | 77.0 | ||
| Stage 4 | 618 | 90.1 | ||
| Overall | 2823 | 51.5 | 88.7 | |
| Liver/bile-duct | ||||
| Stage 1 | 6 | 100.0 | ||
| Stage 2 | 10 | 70.0 | ||
| Stage 3 | 9 | 100.0 | ||
| Stage 4 | 20 | 100.0 | ||
| Overall | 46 | 93.5 | 93.0 | |
| Head & neck | ||||
| Stage 1 | 19 | 63.2 | ||
| Stage 2 | 17 | 82.4 | ||
| Stage 3 | 19 | 84.2 | ||
| Stage 4 | 50 | 96.0 | ||
| Overall | 105 | 85.7 | 93.3 | |
| Esophagus | ||||
| Stage 1 | 8 | 12.5 | ||
| Stage 2 | 17 | 64.7 | ||
| Stage 3 | 34 | 94.1 | ||
| Stage 4 | 40 | 100.0 | ||
| Overall | 100 | 85.0 | - | |
| Pancreatic | ||||
| Stage 1 | 21 | 61.9 | ||
| Stage 2 | 20 | 60.0 | ||
| Stage 3 | 21 | 85.7 | ||
| Stage 4 | 73 | 95.9 | ||
| Overall | 135 | 83.7 | - | |
| Ovary | ||||
| Stage 1 | 10 | 50.0 | ||
| Stage 2 | 5 | 80.0 | ||
| Stage 3 | 31 | 87.1 | ||
| Stage 4 | 19 | 94.7 | ||
| Overall | 65 | 83.1 | 70.4 | |
| Colorectal | ||||
| Stage 1 | 30 | 43.3 | ||
| Stage 2 | 40 | 85.0 | ||
| Stage 3 | 66 | 87.9 | ||
| Stage 4 | 64 | 95.3 | ||
| Overall | 206 | 82.0 | 98.8 | |
| Anus | ||||
| Stage 1 | 4 | 25.0 | ||
| Stage 2 | 4 | 75.0 | ||
| Stage 3 | 13 | 100.0 | ||
| Stage 4 | 1 | 100.0 | ||
| Overall | 22 | 81.8 | 77.8 | |
| Lung | ||||
| Stage 1 | 96 | 21.9 | ||
| Stage 2 | 44 | 79.5 | ||
| Stage 3 | 118 | 90.7 | ||
| Stage 4 | 145 | 95.2 | ||
| Overall | 404 | 74.8 | 91.7 | |
| Plasma cell neoplasm | ||||
| Stage 1 | 17 | 64.7 | ||
| Stage 2 | 16 | 87.5 | ||
| Stage 3 | 14 | 64.3 | ||
| Stage 4 | - | - | ||
| Overall | 47 | 72.3 | - | |
| Stomach | ||||
| Stage 1 | 6 | 16.7 | ||
| Stage 2 | 6 | 50.0 | ||
| Stage 3 | 5 | 80.0 | ||
| Stage 4 | 12 | 100.0 | ||
| Overall | 30 | 66.7 | - | |
| Lymphoma | ||||
| Stage 1 | 33 | 27.3 | ||
| Stage 2 | 48 | 58.3 | ||
| Stage 3 | 46 | 71.7 | ||
| Stage 4 | 46 | 60.9 | ||
| Overall | 174 | 56.3 | - | |
| Bladder | ||||
| Stage 1 | 6 | 33.3 | ||
| Stage 2 | 11 | 9.1 | ||
| Stage 3 | 4 | 75.0 | ||
| Stage 4 | 2 | 100.0 | ||
| Overall | 23 | 34.8 | 87.5 | |
| Unknown primary | ||||
| Stage 1 | - | - | ||
| Stage 2 | 1 | 100.0 | ||
| Stage 3 | 2 | 50.0 | ||
| Stage 4 | 13 | 100.0 | ||
| Overall | 18 | 94.4 | - | |
| Multiple primaries | ||||
| Stage 1 | 2 | 100.0 | ||
| Stage 2 | 5 | 60.0 | ||
| Stage 3 | 6 | 100.0 | ||
| Stage 4 | 6 | 83.3 | ||
| Overall | 19 | 84.2 | - | |
| Urothelial track | ||||
| Stage 1 | 2 | 0.0 | ||
| Stage 2 | - | - | ||
| Stage 3 | - | - | ||
| Stage 4 | 8 | 100.0 | ||
| Overall | 10 | 80.0 | - | |
| Cervix | ||||
| Stage 1 | 12 | 58.3 | ||
| Stage 2 | 5 | 100.0 | ||
| Stage 3 | 7 | 100.0 | ||
| Stage 4 | 1 | 100.0 | ||
| Overall | 25 | 80.0 | 35.0 | |
| Gallbladder | ||||
| Stage 1 | 2 | 0.0 | ||
| Stage 2 | 3 | 33.3 | ||
| Stage 3 | 4 | 75.0 | ||
| Stage 4 | 8 | 100.0 | ||
| Overall | 17 | 70.6 | - | |
| Sarcoma | ||||
| Stage 1 | 10 | 40.0 | ||
| Stage 2 | 2 | 100.0 | ||
| Stage 3 | 10 | 50.0 | ||
| Stage 4 | 7 | 85.7 | ||
| Overall | 30 | 60.0 | - | |
| Other | ||||
| Stage 1 | 11 | 18.2 | ||
| Stage 2 | 3 | 100.0 | ||
| Stage 3 | 18 | 72.7 | ||
| Stage 4 | 18 | 61.1 | ||
| Overall | 59 | 50.8 | - | |
| Melanoma | ||||
| Stage 1 | 2 | 0.0 | ||
| Stage 2 | 2 | 0.0 | ||
| Stage 3 | 3 | 0.0 | ||
| Stage 4 | 6 | 100.0 | ||
| Overall | 13 | 46.2 | 100.0 | |
| Lymphoid leukemia | ||||
| Stage 1 | - | - | ||
| Stage 2 | - | - | ||
| Stage 3 | - | - | ||
| Stage 4 | - | - | ||
| Overall | 51 | 41.2 | - | |
| Breast | ||||
| Stage 1 | 265 | 2.6 | ||
| Stage 2 | 181 | 47.5 | ||
| Stage 3 | 55 | 85.5 | ||
| Stage 4 | 22 | 90.9 | ||
| Overall | 524 | 30.5 | 96.9 | |
| Uterus | ||||
| Stage 1 | 120 | 16.7 | ||
| Stage 2 | 10 | 30.0 | ||
| Stage 3 | 23 | 73.9 | ||
| Stage 4 | 4 | 100.0 | ||
| Overall | 157 | 28.0 | - | |
| Myeloid neoplasm | ||||
| Stage 1 | - | - | ||
| Stage 2 | - | - | ||
| Stage 3 | - | - | ||
| Stage 4 | - | - | ||
| Overall | 10 | 20.0 | - | |
| Kidney | ||||
| Stage 1 | 61 | 4.9 | ||
| Stage 2 | 9 | 22.2 | ||
| Stage 3 | 7 | 14.3 | ||
| Stage 4 | 22 | 54.5 | ||
| Overall | 99 | 18.2 | 77.78 | |
| Prostate | ||||
| Stage 1 | 95 | 3.2 | ||
| Stage 2 | 243 | 4.9 | ||
| Stage 3 | 50 | 14.0 | ||
| Stage 4 | 30 | 83.3 | ||
| Overall | 420 | 11.2 | - | |
| Thyroid | ||||
| Stage 1 | 11 | 0.0 | ||
| Stage 2 | 1 | 0.0 | ||
| Stage 3 | 1 | 0.0 | ||
| Stage 4 | 1 | 0.0 | ||
| Overall | 14 | 0.0 | - |
| Epidemiologic Data for the Australian Population (2010–2014)—Restricted to the Aged Group 55–64 Years [76] | DEEPGEN™ at 95%/99% Specificity * 3604 (n/100,000) | CancerSEEK a at 99.14% Specificity * 1981 (n/100,000) | PanSEER a at 96.1% Specificity * 560 (n/100,000) | GRAIL b at 99.52% Specificity * 4716.1 (n/100,000) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cancer Type | 5-Year Prevalence Rate (n/100,000) | Sensitivity (%) | PPV (%) | Sensitivity (%) | PPV (%) | Sensitivity c (%) | PPV (%) | Sensitivity (%) | PPV (%) |
| Overall | * Specific value for each assay | 57.0/43.0 | 29.9/61.7 | 62.3 | >59.4 | 94.9 | >12.1 | 51.5 | >84.2 |
| Bladder | 40.3 | 32.0/80.0 | 0.6/1.3 | - | - | - | - | 34.8 | 2.8 |
| Brain | 23.0 | - | - | - | - | - | - | - | - |
| Breast (female only) | 1319.4 | 16.0/30.0 | 7.4/17.6 | 33.5 | 34.3 | - | - | 30.5 | 45.9 |
| Primary unknown | 22.3 | - | - | - | - | - | - | 99.4 | 4.4 |
| Cervical | 0.0 | - | - | - | - | - | - | 80.0 | 5.4 |
| Colorectal | 367.9 | 42.0/27.0 | 3.0/9.1 | 64.9 | 21.8 | n.s | - | 82.0 | 38.7 |
| Head and neck | 163.3 | - | - | - | - | - | 85.7 | 22.6 | |
| Liver | 38.7 | 63.0/41.0 | 0.5/1.6 | 98.7 | 4.3 | n.s | - | 93.5 | 7.0 |
| Lung | 132.9 | 67.0/53.0 | 1.8/6.6 | 58.7 | 8.3 | n.s | - | 74.8 | 17.2 |
| Melanoma | 419.8 | - | - | - | - | - | - | 46.2 | 28.9 |
| Non-Hodgkin lymphoma | 145.3 | - | - | - | - | - | - | 56.3 | 14.6 |
| Oesophageal | 20.5 | - | - | 68.1 | 1.6 | n.s | - | 85.0 | 3.5 |
| Ovarian (female only) | 73.2 | - | - | 98.1 | 7.7 | - | - | 83.1 | 11.3 |
| Pancreatic | 28.4 | 52.0/38.0 | 0.3/1.1 | 72 | 2.3 | - | - | 83.7 | 4.7 |
| Prostate (male only) | 1676.4 | 72.0/62.0 | 19.7/51.4 | - | - | - | - | 11.2 | 28.5 |
| Uterine (female only) | 233.3 | - | - | - | - | - | - | 28.0 | 12.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constantin, N.; Sina, A.A.I.; Korbie, D.; Trau, M. Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies. Epigenomes 2022, 6, 6. https://doi.org/10.3390/epigenomes6010006
Constantin N, Sina AAI, Korbie D, Trau M. Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies. Epigenomes. 2022; 6(1):6. https://doi.org/10.3390/epigenomes6010006
Chicago/Turabian StyleConstantin, Nicolas, Abu Ali Ibn Sina, Darren Korbie, and Matt Trau. 2022. "Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies" Epigenomes 6, no. 1: 6. https://doi.org/10.3390/epigenomes6010006
APA StyleConstantin, N., Sina, A. A. I., Korbie, D., & Trau, M. (2022). Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies. Epigenomes, 6(1), 6. https://doi.org/10.3390/epigenomes6010006