
Biochemical Principles in Prion-Based Inheritance


Abstract
1. Introduction
2. Structural Motifs and Amino Acid Sequence Features Observed in Prion Proteins
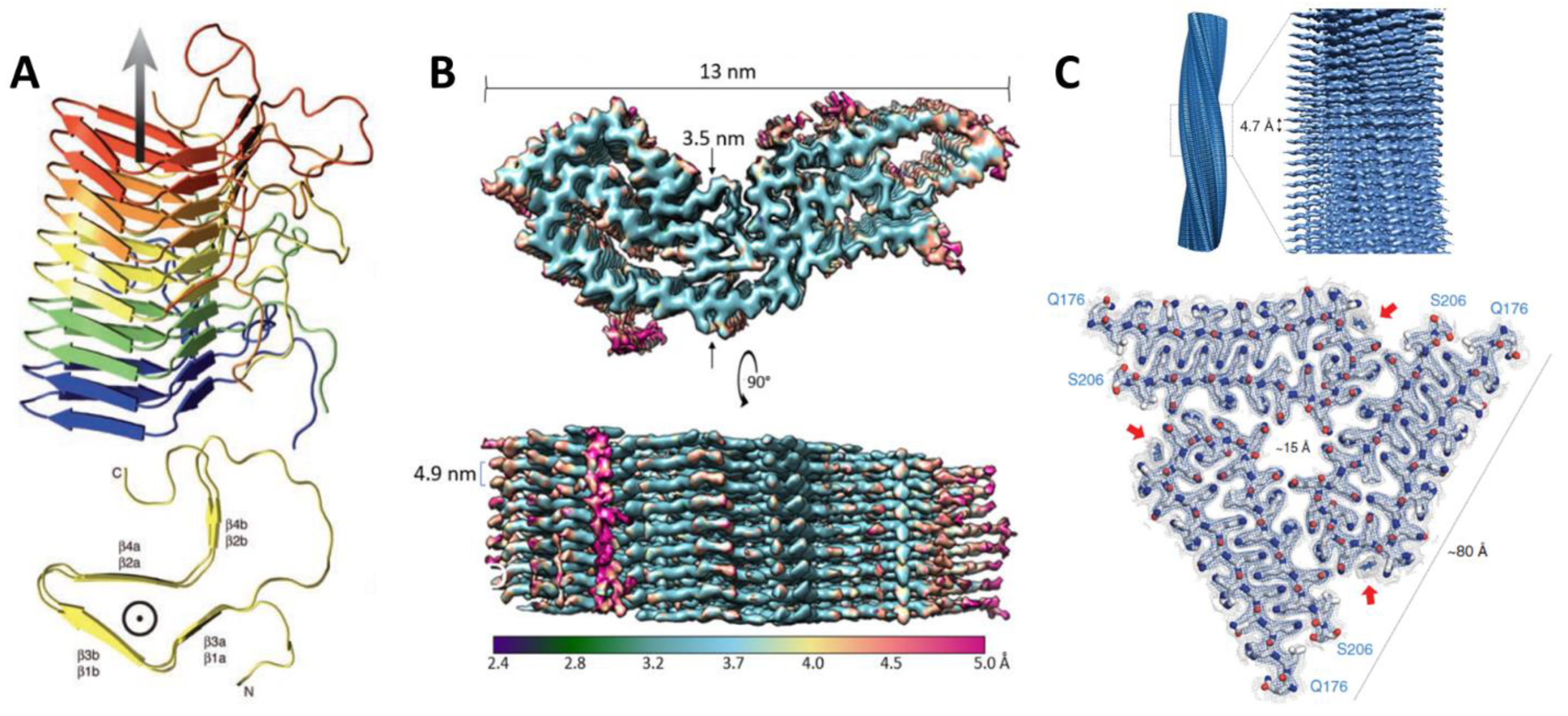
3. High-Resolution Structures of Prions or Prion-like Proteins
4. Conformational Switching, Self-Templating, and Factors Involved in Prion Propagation
5. Contrasting Amyloid-Forming vs. Non-Amyloid Prions, and Methods to Identify Them
6. Conclusions and Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cox, B.S. Ψ, a Cytoplasmic Suppressor of Super-Suppressor in Yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.S.; Lindquist, S. A Heritable Switch in Carbon Source Utilization Driven by an Unusual Yeast Prion. Genes Dev. 2009, 23, 2320–2332. [Google Scholar] [CrossRef]
- Garcia, D.M.; Campbell, E.A.; Jakobson, C.M.; Tsuchiya, M.; Shaw, E.A.; DiNardo, A.L.; Kaeberlein, M.; Jarosz, D.F. A Prion Accelerates Proliferation at the Expense of Lifespan. eLife 2021, 10, e60917. [Google Scholar] [CrossRef]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A Yeast Prion, Mod5, Promotes Acquired Drug Resistance and Cell Survival Under Environmental Stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 2016, 167, 369–381.e12. [Google Scholar] [CrossRef]
- Balbirnie, M.; Grothe, R.; Eisenberg, D.S. An Amyloid-Forming Peptide from the Yeast Prion Sup35 Reveals a Dehydrated β-Sheet Structure for Amyloid. Proc. Natl. Acad. Sci. USA 2001, 98, 2375–2380. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic Structure and Hierarchical Assembly of a Cross-β Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef]
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.J.; Lindquist, S. Self-Seeded Fibers Formed by Sup35, the Protein Determinant of [PSI+], a Heritable Prion-like Factor of S. Cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef]
- King, C.-Y.; Tittmann, P.; Gross, H.; Gebert, R.; Aebi, M.; Wüthrich, K. Prion-Inducing Domain 2–114 of Yeast Sup35 Protein Transforms in Vitro into Amyloid-like Filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 6618–6622. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.M.; Chapman, M.R. Curli Biogenesis and Function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.A.; Quansah, E.; Brundin, P. The Concept of Alpha-Synuclein as a Prion-like Protein: Ten Years After. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Kitamoto, T.; Tateishi, J.; Tashima, T.; Takeshita, I.; Barry, R.A.; DeArmond, S.J.; Prusiner, S.B. Amyloid Plaques in Creutzfeldt-Jakob Disease Stain with Prion Protein Antibodies. Ann. Neurol. 1986, 20, 204–208. [Google Scholar] [CrossRef]
- Garcia, D.M.; Jarosz, D.F. Rebels with a Cause: Molecular Features and Physiological Consequences of Yeast Prions. FEMS Yeast Res. 2014, 14, 136–147. [Google Scholar] [CrossRef]
- Nan, H.; Chen, H.; Tuite, M.F.; Xu, X. A Viral Expression Factor Behaves as a Prion. Nat. Commun. 2019, 10, 359. [Google Scholar] [CrossRef]
- Yuan, A.H.; Hochschild, A. A Bacterial Global Regulator Forms a Prion. Science 2017, 355, 198–201. [Google Scholar] [CrossRef]
- Holmes, D.L.; Lancaster, A.K.; Lindquist, S.; Halfmann, R. Heritable Remodeling of Yeast Multicellularity by an Environmentally Responsive Prion. Cell 2013, 153, 153–165. [Google Scholar] [CrossRef]
- True, H.L.; Lindquist, S.L. A Yeast Prion Provides a Mechanism for Genetic Variation and Phenotypic Diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.K.; Smejkal, T.; Itakura, A.K.; Garcia, D.M.; Jarosz, D.F. A Non-Amyloid Prion Particle That Activates a Heritable Gene Expression Program. Mol. Cell 2020, 77, 251–265.e9. [Google Scholar] [CrossRef] [PubMed]
- Harvey, Z.H.; Chakravarty, A.K.; Futia, R.A.; Jarosz, D.F. A Prion Epigenetic Switch Establishes an Active Chromatin State. Cell 2020, 180, 928–940.e14. [Google Scholar] [CrossRef]
- Lacroute, F. Non-Mendelian Mutation Allowing Ureidosuccinic Acid Uptake in Yeast. J. Bacteriol. 1971, 106, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. [URE3] as an Altered URE2 Protein: Evidence for a Prion Analog in Saccharomyces Cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and Environmental Factors Affecting the De Novo Appearance of the [Psi(+)] Prion in Saccharomyces Cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions Affect the Appearance of Other Prions: The Story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The Protein Product of the Het-s Heterokaryon Incompatibility Gene of the Fungus Podospora Anserina Behaves as a Prion Analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef]
- Du, Z.; Park, K.-W.; Yu, H.; Fan, Q.; Li, L. Newly Identified Prion Linked to the Chromatin-Remodeling Factor Swi1 in Saccharomyces Cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef]
- Patel, B.K.; Gavin-Smyth, J.; Liebman, S.W. The Yeast Global Transcriptional Co-Repressor Protein Cyc8 Can Propagate as a Prion. Nat. Cell Biol. 2009, 11, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Rogoza, T.; Goginashvili, A.; Rodionova, S.; Ivanov, M.; Viktorovskaya, O.; Rubel, A.; Volkov, K.; Mironova, L. Non-Mendelian Determinant [ISP+] in Yeast Is a Nuclear-Residing Prion Form of the Global Transcriptional Regulator Sfp1. Proc. Natl. Acad. Sci. USA 2010, 107, 10573–10577. [Google Scholar] [CrossRef] [PubMed]
- Volkov, K.V.; Aksenova, A.Y.; Soom, M.J.; Osipov, K.V.; Svitin, A.V.; Kurischko, C.; Shkundina, I.S.; Ter-Avanesyan, M.D.; Inge-Vechtomov, S.G.; Mironova, L.N. Novel Non-Mendelian Determinant Involved in the Control of Translation Accuracy in Saccharomyces cerevisiae. Genetics 2002, 160, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Saifitdinova, A.F.; Nizhnikov, A.A.; Lada, A.G.; Rubel, A.A.; Magomedova, Z.M.; Ignatova, V.V.; Inge-Vechtomov, S.G.; Galkin, A.P. [NSI+]: A Novel Non-Mendelian Nonsense Suppressor Determinant in Saccharomyces Cerevisiae. Curr. Genet. 2010, 56, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why Are? Natively Unfolded? Proteins Unstructured under Physiologic Conditions? Proteins Struct. Funct. Genet. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Li, L.; Lindquist, S. Creating a Protein-Based Element of Inheritance. Science 2000, 287, 661–664. [Google Scholar] [CrossRef]
- Stein, K.C.; True, H.L. Extensive Diversity of Prion Strains Is Defined by Differential Chaperone Interactions and Distinct Amyloidogenic Regions. PLoS Genet. 2014, 10, e1004337. [Google Scholar] [CrossRef]
- Ba, A.N.N.; Yeh, B.J.; van Dyk, D.; Davidson, A.R.; Andrews, B.J.; Weiss, E.L.; Moses, A.M. Proteome-Wide Discovery of Evolutionary Conserved Sequences in Disordered Regions. Sci. Signal. 2012, 5, rs1. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Uversky, V.N.; Dunker, A.K.; Kurgan, L. Chapter 1—Introduction to Intrinsically Disordered Proteins and Regions. In Intrinsically Disordered Proteins; Salvi, N., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–34. ISBN 978-0-12-816348-1. [Google Scholar]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM Structure of a Neuronal Functional Amyloid Implicated in Memory Persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Van Melckebeke, H.; Wasmer, C.; Lange, A.; Ab, E.; Loquet, A.; Böckmann, A.; Meier, B.H. Atomic-Resolution Three-Dimensional Structure of HET-s(218-289) Amyloid Fibrils by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 13765–13775. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid Fibrils of the HET-s(218–289) Prion Form a β Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-Resolution Structure and Strain Comparison of Infectious Mammalian Prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef] [PubMed]
- Amenitsch, H.; Benetti, F.; Ramos, A.; Legname, G.; Requena, J.R. SAXS Structural Study of PrP(Sc) Reveals ~11 Nm Diameter of Basic Double Intertwined Fibers. Prion 2013, 7, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Sim, V.L.; Caughey, B. Ultrastructures and Strain Comparison of Under-Glycosylated Scrapie Prion Fibrils. Neurobiol. Aging 2009, 30, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full Atomistic Model of Prion Structure and Conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef] [PubMed]
- Terry, C.; Harniman, R.L.; Sells, J.; Wenborn, A.; Joiner, S.; Saibil, H.R.; Miles, M.J.; Collinge, J.; Wadsworth, J.D.F. Structural Features Distinguishing Infectious Ex Vivo Mammalian Prions from Non-Infectious Fibrillar Assemblies Generated In Vitro. Sci. Rep. 2019, 9, 376. [Google Scholar] [CrossRef]
- Vázquez-Fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef]
- Si, K.; Kandel, E.R. The Role of Functional Prion-Like Proteins in the Persistence of Memory. Cold Spring Harb. Perspect. Biol. 2016, 8, a021774. [Google Scholar] [CrossRef]
- Jackson, G.S.; Hosszu, L.L.P.; Power, A.; Hill, A.F.; Kenney, J.; Saibil, H.; Craven, C.J.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Reversible Conversion of Monomeric Human Prion Protein Between Native and Fibrilogenic Conformations. Science 1999, 283, 1935–1937. [Google Scholar] [CrossRef]
- Bradley, M.E.; Liebman, S.W. The Sup35 Domains Required for Maintenance of Weak, Strong or Undifferentiated Yeast [PSI+] Prions. Mol. Microbiol. 2004, 51, 1649–1659. [Google Scholar] [CrossRef]
- Collins, S.R.; Douglass, A.; Vale, R.D.; Weissman, J.S. Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol. 2004, 2, e321. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Lindquist, S.L. Structural Insights into a Yeast Prion Illuminate Nucleation and Strain Diversity. Nature 2005, 435, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Tessier, P.M.; Lindquist, S. Prion Recognition Elements Govern Nucleation, Strain Specificity and Species Barriers. Nature 2007, 447, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Villali, J.; Dark, J.; Brechtel, T.M.; Pei, F.; Sindi, S.S.; Serio, T.R. Nucleation Seed Size Determines Amyloid Clearance and Establishes a Barrier to Prion Appearance in Yeast. Nat. Struct. Mol. Biol. 2020, 27, 540–549. [Google Scholar] [CrossRef]
- Keefer, K.M.; Stein, K.C.; True, H.L. Heterologous Prion-Forming Proteins Interact to Cross-Seed Aggregation in Saccharomyces Cerevisiae. Sci. Rep. 2017, 7, 5853. [Google Scholar] [CrossRef]
- Serio, T.R. [PIN+]Ing down the Mechanism of Prion Appearance. FEMS Yeast Res. 2018, 18, foy026. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the Chaperone Protein Hsp104 in Propagation of the Yeast Prion-Like Factor [ Psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Hsp104 Catalyzes Formation and Elimination of Self-Replicating Sup35 Prion Conformers. Science 2004, 304, 1793–1797. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Destruction or Potentiation of Different Prions Catalyzed by Similar Hsp104 Remodeling Activities. Mol. Cell 2006, 23, 425–438. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Hsp104, Hsp70 and Hsp40 Interplay Regulates Formation, Growth and Elimination of Sup35 Prions. EMBO J. 2008, 27, 2712–2724. [Google Scholar] [CrossRef]
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among Prions and Prion “Strains” in Yeast. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16392–16399. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and Variability of [Ps1] Prion Factors in Saccharomyces Cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational Variations in an Infectious Protein Determine Prion Strain Differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Schlumpberger, M.; Prusiner, S.B.; Herskowitz, I. Induction of Distinct [URE3] Yeast Prion Strains. Mol. Cell. Biol. 2001, 21, 7035–7046. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Son, M.; Edskes, H.K. Prion Variants of Yeast Are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems. Viruses 2019, 11, 238. [Google Scholar] [CrossRef]
- Derdowski, A.; Sindi, S.S.; Klaips, C.L.; DiSalvo, S.; Serio, T.R. A Size Threshold Limits Prion Transmission and Establishes Phenotypic Diversity. Science 2010, 330, 680–683. [Google Scholar] [CrossRef]
- Alberti, S. Phase Separation in Biology. Curr. Biol. 2017, 27, R1097–R1102. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase Separation of a Yeast Prion Protein Promotes Cellular Fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef]
- Toyama, B.H.; Kelly, M.J.S.; Gross, J.D.; Weissman, J.S. The Structural Basis of Yeast Prion Strain Variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] Prion Aggregates Are Formed by Small Sup35 Polymers Fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Dergalev, A.A.; Alexandrov, A.I. Proteinase K Resistant Cores of Prions and Amyloids. Prion 2020, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an Amyloid Dye: Fibril Quantification, Optimal Concentration and Effect on Aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and Amyloids: History and Relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Lindquist, S. Screening for Amyloid Aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J. Vis. Exp. 2008. [Google Scholar] [CrossRef] [PubMed]
- Hanna-Addams, S.; Wang, Z. Use of Two Dimensional Semi-Denaturing Detergent Agarose Gel Electrophoresis to Confirm Size Heterogeneity of Amyloid or Amyloid-like Fibers. J. Vis. Exp. JoVE 2018, 134, 57498. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Jarosz, D.F.; Jones, S.K.; Chang, A.; Lancaster, A.K.; Lindquist, S. Prions Are a Common Mechanism for Phenotypic Inheritance in Wild Yeasts. Nature 2012, 482, 363–368. [Google Scholar] [CrossRef]
- Greene, L.; Park, Y.-N.; Masison, D.; Eisenberg, E. Application of GFP-Labeling to Study Prions in Yeast. Protein Pept. Lett. 2009, 16, 635–641. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patino, M.M.; Liu, J.-J.; Glover, J.R.; Lindquist, S. Support for the Prion Hypothesis for Inheritance of a Phenotypic Trait in Yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef]
- Khan, T.; Kandola, T.S.; Wu, J.; Venkatesan, S.; Ketter, E.; Lange, J.J.; Rodríguez Gama, A.; Box, A.; Unruh, J.R.; Cook, M.; et al. Quantifying Nucleation In Vivo Reveals the Physical Basis of Prion-like Phase Behavior. Mol. Cell 2018, 71, 155–168. [Google Scholar] [CrossRef]
- Newby, G.A.; Kiriakov, S.; Hallacli, E.; Kayatekin, C.; Tsvetkov, P.; Mancuso, C.P.; Bonner, J.M.; Hesse, W.R.; Chakrabortee, S.; Manogaran, A.L.; et al. A Genetic Tool to Track Protein Aggregates and Control Prion Inheritance. Cell 2017, 171, 966–979.e18. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.W.; Bagriantsev, S.N.; Derkatch, I.L. Biochemical and Genetic Methods for Characterization of [PIN+] Prions in Yeast. Methods 2006, 39, 23–34. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Amyloid-Forming Prions | Non-Amyloid-Forming Prions |
|---|---|
| PrPSc [2] 1 [URE3] [25,26] [PSI+] [1,26] [RNQ+]/[PIN+] [27,28] [MOT3+] [3] [MOD+] [6] [Het-s] [29] 2 [SWI+] [30] [OCT+] [31] [ISP+] [32,33] [NSI+] [34] Cb-Rho [20] 3 | [GAR+] [4] [SMAUG+] [23] [ESI+] [24] [BIG+] [5] Others [7] |
| Prion/Protein | [Het-s] | PrPSc | Orb2B |
|---|---|---|---|
| Gene name | het-s | PRNP | orb2 |
| Core residues (aa) | 218–289 (72) | 95–227 (133) | 176–206 (31) |
| Structure/Symmetry | Left-handed solenoid | Parallel in-register β-sheets | Threefold triangular symmetry |
| Method to interpret structure | ss-NMR | Cryo-EM | Cryo-EM |
| Core | Hydrophobic | Hydrophobic | Hydrophilic |
| Stabilizing features | 23 hydrogen bonds, three salt bridges, two asparagine ladders | GPI anchor, “Greek Key” motif, β-arches | Interdigitated cross-β structure, protonation of histidine |
| Post-translational modifications | Unknown | N-linked glycosylation | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dennis, E.M.; Garcia, D.M. Biochemical Principles in Prion-Based Inheritance. Epigenomes 2022, 6, 4. https://doi.org/10.3390/epigenomes6010004
Dennis EM, Garcia DM. Biochemical Principles in Prion-Based Inheritance. Epigenomes. 2022; 6(1):4. https://doi.org/10.3390/epigenomes6010004
Chicago/Turabian StyleDennis, Emily M., and David M. Garcia. 2022. "Biochemical Principles in Prion-Based Inheritance" Epigenomes 6, no. 1: 4. https://doi.org/10.3390/epigenomes6010004
APA StyleDennis, E. M., & Garcia, D. M. (2022). Biochemical Principles in Prion-Based Inheritance. Epigenomes, 6(1), 4. https://doi.org/10.3390/epigenomes6010004