
SWI/SNF Chromatin Remodeling Enzymes in Melanoma



Abstract
:1. Introduction
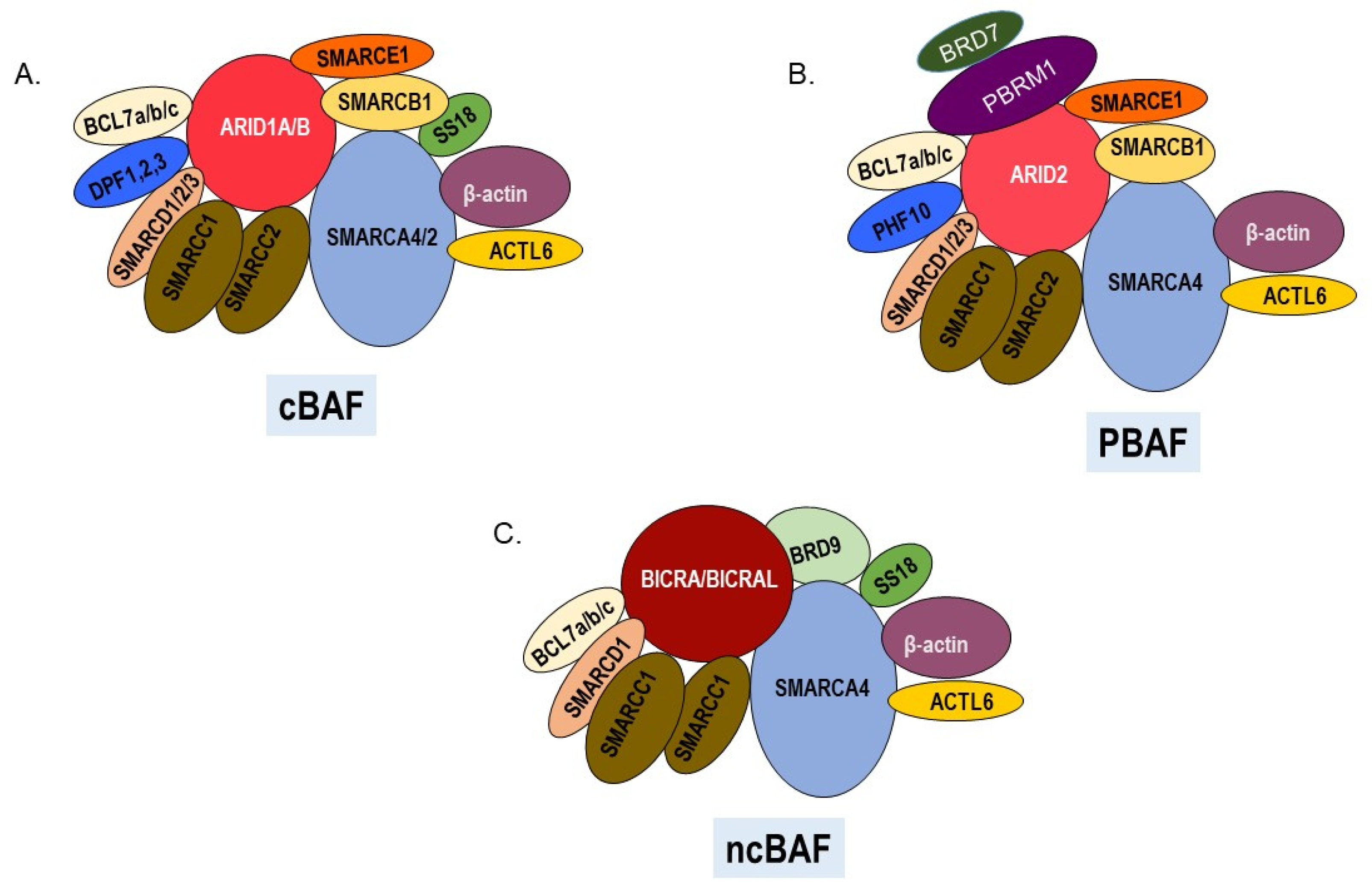
1.1. SWI/SNF Chromatin Remodeling Complexes
1.2. Melanoma
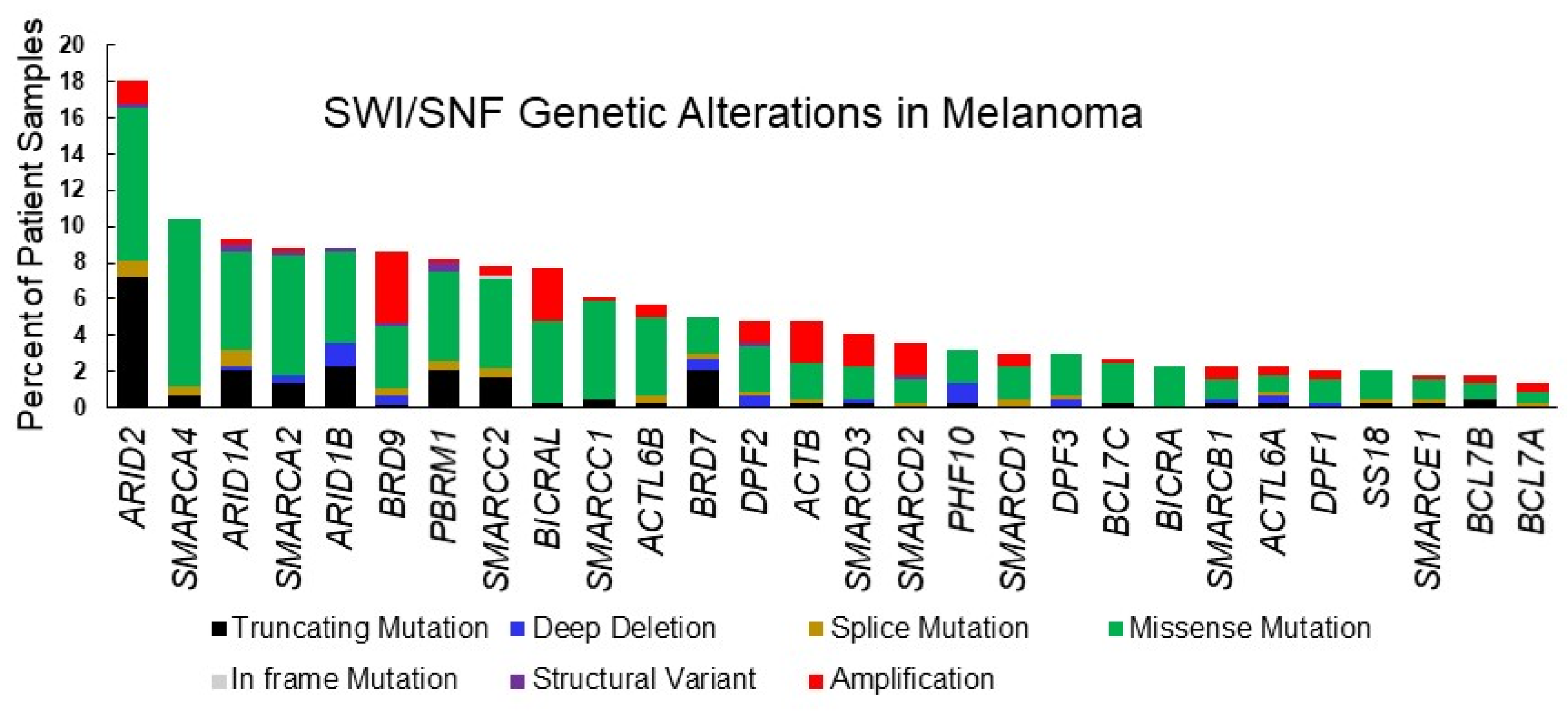
2. SWI/SNF in Melanoma
2.1. SMARCA4
2.2. SMARCA2 (BRM)
2.3. SMARCB1 (INI1/BAF47/SNF5)
2.4. SMARCD1, 2, 3 (BAF60A, B, C)
2.5. ARID1A andARID1B
2.6. ARID2 and Other Components of the PBAF Complex

{kind=link}
{kind=link}
{kind=link}
| Subunit | Function in SWI/SNF | General Cellular Functions | Specific Functions in Melanocytes/Melanoma |
|---|---|---|---|
| SMARCA4 | Central ATPase in PBAF, ncBAF, and a subset of cBAF complexes; also has a bromodomain [5,6,34]. | Required for mouse development [68], embryonic stem cell pluripotency [26], promotes nucleotide excision repair [85]. | Has ambivalent roles with some reports indicating low expression [73] and others high expression [74,75,76]. Required for melanocyte development, melanoma tumorigenicity, co-activator for MITF and SOX10, promotes melanin synthesis, increases resistance to DNA-damaging agents [47,75,77,78,79,80]. Promotes tumorigenesis in BRAFV600E-driven mouse models [89]. Suppresses tumorigenesis in orthotopic models of melanoma [90]. |
| SMARCA2 | Central ATPase in a subset of cBAF complexes; also has a bromodomain [5,6,34]. | High frequency of mutations in sun-exposed non-melanoma skin cancers [93]. Expression can be suppressed by oncogenes and activity inhibited by acetylation [95,96]. Synthetic lethal with SMARCA4 [98]. | Interacts with MITF and compensates for SMARCA4 loss in some melanoma cells [77]. Associated with human variation in pigmentation [92] and with senescent melanocytes [94]. |
| SMARCB1 | Core component of cBAF and PBAF complexes. Interacts with the acidic patch of the nucleosome [18,19,20,21]. | Homozygous disruption is embryonic lethal; mice with heterozygous disruption develop tumors with loss of heterozygosity [110,111]. Involved in nucleotide excision repair [112]. | Has ambivalent roles. May be required for mutant BRAF-induced senescence [114]. Loss also results in senescence, increasing sensitivity to BCL2 inhibitors and resistance to BRAF inhibitors [116]. |
| SMARCD1 | Component of ncBAF and a subset of cBAF and PBAF complexes [5,6]. | Associated with embryonic stem cell self-renewal and pluripotency, bivalent marks, nuclear hormone, p53, SOX10 (Schwann cell) interactions [29,32,117,118,123]. | Interacts with MITF and SOX10 in melanocytes and melanoma cells [79,122]. |
| SMARCD2 | Component of a subset of cBAF and PBAF complexes [5,6] | Involved in neutrophil differentiation, interacts with p53 and ATM to preserve cell identity [119,120,121]. | Interacts with MITF in melanocytes and melanoma cells [79,122]. |
| SMARCD3 | Component of a subset of cBAF and PBAF complexes [5,6]. | Required for muscle differentiation [124]. Involved in glycolytic metabolism and lipogenesis [125,126]. | Correlates with poorer patient survival in uveal melanoma [127]. |
| ARID1A | Component of some cBAF complexes. Has important function in determining SWI/SNF architecture and ability to mobilize nucleosomes [19,20,21]. | Most frequently mutated SWI/SNF gene in cancer [128]. Promotes expression of interferon γ-regulated genes [132]. Associated with lineage-specific enhancers [22,24]. | Mutations associated with late stages and EZH2 program. Melanoma patients with tumors that have high levels correlate with better response to immune checkpoint inhibitors [132]. |
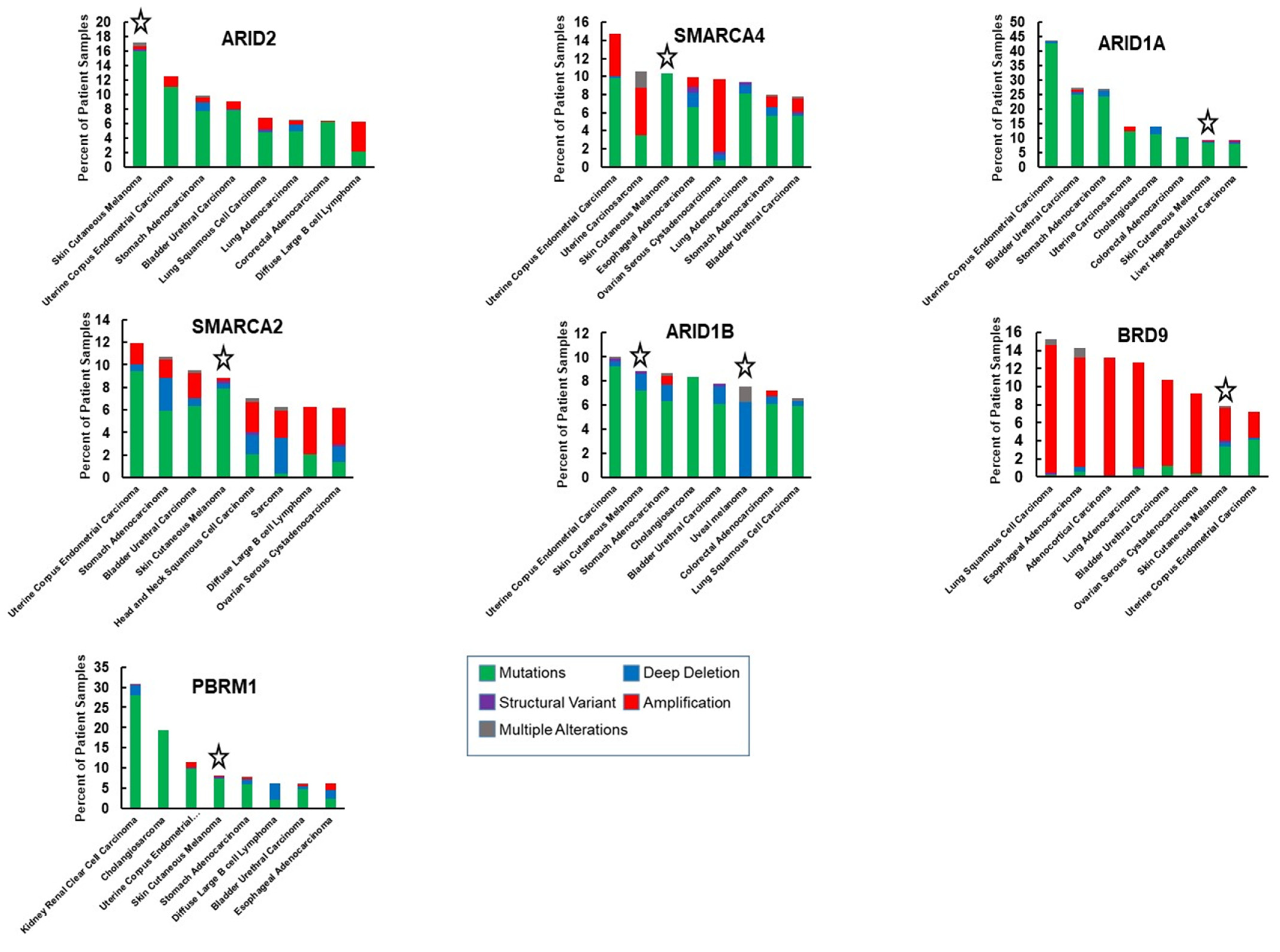
| ARID1B | Component of a subset of cBAF complexes [5,6]. | Associated with lineage-specific enhancer activation [22,24]. Compensates for ARID1A loss in some cancers and is synthetic lethal with ARID1A loss [24,143]. Dual loss of ARID1A/ARID1B can also be pro-tumorigenic [144]. | High frequency of copy-number losses in mucosal melanomas [141]. High frequency of deep deletion in uveal melanoma (Figure 3). |
| ARID2 | Component of PBAF complexes [5,6]. | Functions in DNA repair and genome integrity [146,147]. Occupies and activates lineage-specific enhancers during osteogenesis [170]. | Mutations are associated with UVR exposure and coincide with the transition to melanoma in situ [130]. Suppresses invasion in vitro and modulates response to immunotherapy in vivo [150,152]. |
| PBRM1 | Component of PBAF complexes that has six tandem bromodomains [5,6,34]. | Frequently mutated in renal clear cell carcinoma 153]. Loss is synthetic lethal with inhibitors of DNA repair [157]. | Component of MITF interactome [79]. Modulates response to immunotherapy by regulating interferon γ inducible genes [152]. |
| BRD7 | Bromodomain-containing component of PBAF complexes [5,6,34]. | Positive regulator of p53-induced senescence [158,159,160]; also interacts with MYC, promotes colorectal cancer growth and is associated with poorer prognosis in multiple myeloma [162,163]. | High expression was associated with poorer patient survival and anti-tumorigenic response obtained with TP-772 [161]. |
| PHF10 | Component of PBAF complexes [5,6]. | In Drosophila, involved in transcriptional elongation [164]. Activates NF-kβ target genes [165]. Promotes proliferation of gastric cancer cells [168,169]. | Homozygous deletion and frame-shift mutations in uveal melanoma [166]. Over-expressed in cutaneous melanoma and interacts with MYC to promote proliferation [167]. |
| BRD9 | Bromodomain-containing component of ncBAF complexes [5,6,34]. | Vulnerability in cancers with SMARCB1 inactivation [171] and tumors with SS18-SSX fusion [172]. Inhibition of BRD9 suppresses tumorigenicity of diverse cancers [37,38,173,174]. | Ambivalent role in melanoma. Over-expressed in melanoma and associated with the anti-tumorigenic response to TP-772 [161]. Expression is lost in uveal melanoma due to mis-splicing and incorporation of a poison exon as a result of mutations in SF3B1 [90]. |
2.7. BRD9 and ncBAF
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Winston, F.; Carlson, M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992, 8, 387–391. [Google Scholar] [CrossRef]
- Middeljans, E.; Wan, X.; Jansen, P.W.; Sharma, V.; Stunnenberg, H.G.; Logie, C. SS18 Together with Animal-Specific Factors Defines Human BAF-Type SWI/SNF Complexes. PLoS ONE 2012, 7, e33834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashtalir, N.; D’Avino, A.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; Pierre, R.S.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innis, S.M.; Cabot, B. GBAF, a small BAF sub-complex with big implications: A systematic review. Epigenet. Chromatin 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- De La Serna, I.L.; Ohkawa, Y.; Imbalzano, A.N. Chromatin remodelling in mammalian differentiation: Lessons from ATP-dependent remodellers. Nat. Rev. Genet. 2006, 7, 461–473. [Google Scholar] [CrossRef]
- Alfert, A.; Moreno, N.; Kerl, K. The BAF complex in development and disease. Epigenet. Chromatin 2019, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar] [PubMed]
- Ramos, P.; Karnezis, A.N.; Craig, D.W.; Sekulic, A.; Russell, M.L.; Hendricks, W.P.; Corneveaux, J.J.; Barrett, M.T.; Shumansky, K.; Yang, Y.; et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, K.C.; Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Ben Maad, I.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a Core Chromatin Remodeling Complex from SWI/SNF Subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Han, Y.; Reyes, A.A.; Malik, S.; He, Y. Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature 2020, 579, 452–455. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef]
- Mashtalir, N.; Suzuki, H.; Farrell, D.P.; Sankar, A.; Luo, J.; Filipovski, M.; D’Avino, A.R.; Pierre, R.S.; Valencia, A.M.; Onikubo, T.; et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 2020, 183, 802–817.e24. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alver, B.; Roman, R.M.A.K.S.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, B.H.A.P.J.; Shivdasani, A.A.K.S.R.R.; Roberts, R.M.B.G.W.X.W.C.W.M. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tao, T.; Abraham, B.J.; Durbin, A.D.; Zimmerman, M.W.; Kadoch, C.; Look, A.T. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci. Adv. 2020, 6, eaaz3440. [Google Scholar] [CrossRef] [PubMed]
- Kelso, T.; Porter, D.K.; Amaral, M.L.; Shokhirev, M.N.; Benner, C.; Hargreaves, D.C. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 2017, 6, e30506. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Patsialou, A.; Zumbrun, S.D.; Kim, S.; Chen, Y.; Dallas, P.B.; Moran, E. The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 2004, 32, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Saladi, S.V.; de la Serna, I.L. ATP dependent chromatin remodeling enzymes in embryonic stem cells. Stem. Cell Rev. 2010, 6, 62–73. [Google Scholar] [CrossRef] [Green Version]
- de la Serna, I.L.; Ohkawa, Y.; Berkes, C.A.; Bergstrom, D.A.; Dacwag, C.S.; Tapscott, S.J.; Imbalzano, A.N. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol. Cell Biol. 2005, 25, 3997–4009. [Google Scholar] [CrossRef] [Green Version]
- de la Serna, I.L.; Ohkawa, Y.; Higashi, C.; Dutta, C.; Osias, J.; Kommajosyula, N.; Tachibana, T.; Imbalzano, A.N. The microphthalmia-associated transcription factor requires SWI/SNF enzymes to activate melanocyte-specific genes. J. Biol. Chem. 2006, 281, 20233–20241. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, P.-W.; Fryer, C.J.; Trotter, K.W.; Wang, W.; Archer, T.K. BAF60a Mediates Critical Interactions between Nuclear Receptors and the BRG1 Chromatin-Remodeling Complex for Transactivation. Mol. Cell. Biol. 2003, 23, 6210–6220. [Google Scholar] [CrossRef] [Green Version]
- Belandia, B.; Orford, R.L.; Hurst, H.C.; Parker, M.G. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002, 21, 4094–4103. [Google Scholar] [CrossRef]
- Inoue, H.; Furukawa, T.; Giannakopoulos, S.; Zhou, S.; King, D.S.; Tanese, N. Largest Subunits of the Human SWI/SNF Chromatin-remodeling Complex Promote Transcriptional Activation by Steroid Hormone Receptors. J. Biol. Chem. 2002, 277, 41674–41685. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Sohn, D.H.; Ko, M.; Chung, H.; Jeon, S.H.; Seong, R.H. BAF60a Interacts with p53 to Recruit the SWI/SNF Complex. J. Biol. Chem. 2008, 283, 11924–11934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.W.; Davies, K.P.; Yung, E.; Beltran, R.J.; Yu, J.; Kalpana, G.V. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat. Genet. 1999, 22, 102–105. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.-S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, T.; Robinson, D.C.L.; Witwicka, H.; Dilworth, F.J.; Imbalzano, A.N. The Bromodomains of the mammalian SWI/SNF (mSWI/SNF) ATPases Brahma (BRM) and Brahma Related Gene 1 (BRG1) promote chromatin interaction and are critical for skeletal muscle differentiation. Nucleic Acids Res. 2021, 49, 8060–8077. [Google Scholar] [CrossRef]
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.-S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpsoy, A.; Utturkar, S.M.; Carter, B.C.; Dhiman, A.; Torregrosa-Allen, S.E.; Currie, M.P.; Elzey, B.D.; Dykhuizen, E.C. BRD9 Is a Critical Regulator of Androgen Receptor Signaling and Prostate Cancer Progression. Cancer Res. 2021, 81, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Hugle, M.; Regenass, P.; Warstat, R.; Hau, M.; Schmidtkunz, K.; Lucas, X.; Wohlwend, D.; Einsle, O.; Jung, M.; Breit, B.; et al. 4-Acyl Pyrroles as Dual BET-BRD7/9 Bromodomain Inhibitors Address BETi Insensitive Human Cancer Cell Lines. J. Med. Chem. 2020, 63, 15603–15620. [Google Scholar] [CrossRef]
- Imbalzano, A.N.; Kwon, H.; Green, M.R.; Kingston, R.E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994, 370, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, S.R.; Xie, X.; Hosny El Said, N.; Venit, T.; Gunsalus, K.C.; Percipalle, P. beta-actin dependent chromatin remodeling mediates compartment level changes in 3D genome architecture. Nat. Commun. 2021, 12, 5240. [Google Scholar] [CrossRef] [PubMed]
- Rahnamoun, H.; Lee, J.; Sun, Z.; Lu, H.; Ramsey, K.; Komives, E.A.; Lauberth, S.M. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat. Struct. Mol. Biol. 2018, 25, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Raible, F.; Mollaaghababa, R.; Guyon, J.R.; Wu, C.-T.; Bender, W.; Kingston, R.E. Stabilization of Chromatin Structure by PRC1, a Polycomb Complex. Cell 1999, 98, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.; Cho, Y.-J.; Koellhoffer, E.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic Antagonism between Polycomb and SWI/SNF Complexes during Oncogenic Transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saladi, S.; Wong, P.G.; Trivedi, A.R.; Marathe, H.G.; Keenen, B.; Aras, S.; Liew, Z.; Setaluri, V.; La Serna, I.L. BRG 1 promotes survival of UV-irradiated melanoma cells by cooperating with MITF to activate the melanoma inhibitor of apoptosis gene. Pigment. Cell Melanoma Res. 2013, 26, 377–391. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.-J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies–update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Kuperwasser, C.; Brunet, J.-P.; Ramaswamy, S.; Kuo, W.-L.; Gray, J.W.; Naber, S.P.; Weinberg, R.A. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat. Genet. 2005, 37, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the Microphthalmia Transcription Factor Network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Widlund, H.; Rubin, M.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, J.D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Harris, M.L.; Baxter, L.L.; Loftus, S.K.; Pavan, W.J. Sox proteins in melanocyte development and melanoma. Pigment. Cell Melanoma Res. 2010, 23, 496–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronin, J.C.; Watkins-Chow, D.E.; Incao, A.; Hasskamp, J.H.; Schonewolf, N.; Aoude, L.G.; Hayward, N.K.; Bastian, B.C.; Dummer, R.; Loftus, S.K.; et al. SOX10 ablation arrests cell cycle, induces senescence, and suppresses melanomagenesis. Cancer Res. 2013, 73, 5709–5718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Sample, A.; He, Y.-Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef]
- Ottaviano, M.; Giunta, E.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; Dias, S.D.R.; Hayward, R.; Savage, K.; Delmas, V.; LaRue, L.; Pritchard, C.; Marais, R. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Lorusso, P.M.; Schalper, K.; Sosman, J. Targeted therapy and immunotherapy: Emerging biomarkers in metastatic melanoma. Pigment. Cell Melanoma Res. 2020, 33, 390–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaddour, K.; Maahs, L.; Avila-Rodriguez, A.M.; Maamar, Y.; Samaan, S.; Ansstas, G. Melanoma Targeted Therapies beyond BRAF-Mutant Melanoma: Potential Druggable Mutations and Novel Treatment Approaches. Cancers 2021, 13, 5847. [Google Scholar] [CrossRef]
- Shain, A.H.; Pollack, J.R. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 Null Mutation in the Mouse Reveals Functional Differences among Mammalian SWI/SNF Complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Orvis, T.; Hepperla, A.; Walter, V.; Song, S.; Simon, J.; Parker, J.; Wilkerson, M.D.; Desai, N.; Major, M.B.; Hayes, D.N.; et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014, 74, 6486–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Madany, P.; Akech, J.; Dobson, J.R.; Douthwright, S.; Browne, G.; Colby, J.L.; Winter, G.E.; Bradner, J.E.; Pratap, J.; et al. The SWI/SNF ATPases Are Required for Triple Negative Breast Cancer Cell Proliferation. J. Cell. Physiol. 2015, 230, 2683–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.M.; Haferkamp, S.; Dijkstra, M.K.; Scurr, L.L.; Frausto, M.; Diefenbach, E.; Scolyer, R.A.; Reisman, D.N.; Mann, G.J.; Kefford, R.F.; et al. The chromatin remodelling factor BRG1 is a novel binding partner of the tumor suppressor p16INK4a. Mol. Cancer 2009, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Wong, R.P.C.; Martinka, M.; Li, G. BRG1 expression is increased in human cutaneous melanoma. Br. J. Dermatol. 2010, 163, 502–510. [Google Scholar] [CrossRef]
- Saladi, S.V.; Keenen, B.; Marathe, H.G.; Qi, H.; Chin, K.-V.; de la Serna, I.L. Modulation of extracellular matrix/adhesion molecule expression by BRG1 is associated with increased melanoma invasiveness. Mol. Cancer 2010, 9, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Li, J.; Wu, J.; Xu, B.; Wang, Z.; Giamas, G.; Stebbing, J.; Yu, Z. A Pan-Cancer Analysis of SMARCA4 Alterations in Human Cancers. Front. Immunol. 2021, 12, 762598. [Google Scholar] [CrossRef] [PubMed]
- Keenen, B.; Qi, H.; Saladi, S.; Yeung, M.; De La Serna, I. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene 2009, 29, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachtenheim, J.; Ondrušová, L.; Borovanský, J. SWI/SNF chromatin remodeling complex is critical for the expression of microphthalmia-associated transcription factor in melanoma cells. Biochem. Biophys. Res. Commun. 2010, 392, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Laurette, P.; Strub, T.; Koludrovic, D.; Keime, C.; Le Gras, S.; Seberg, H.; Van Otterloo, E.; Imrichova, H.; Siddaway, R.; Aerts, S.; et al. Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. eLife 2015, 4, e06857. [Google Scholar] [CrossRef] [PubMed]
- Marathe, H.G.; Watkins-Chow, D.E.; Weider, M.; Hoffmann, A.; Mehta, G.; Trivedi, A.; Aras, S.; Basuroy, T.; Mehrotra, A.; Bennett, D.; et al. BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation. Nucleic Acids Res. 2017, 45, 6442–6458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, J.S.; Bell, R.J.A.; Tran, T.-N.T.; Haq, R.; Liu, H.; Love, K.T.; Langer, R.; Anderson, D.G.; LaRue, L.; et al. YY1 Regulates Melanocyte Development and Function by Cooperating with MITF. PLoS Genet. 2012, 8, e1002688. [Google Scholar] [CrossRef] [Green Version]
- Seberg, H.E.; Van Otterloo, E.; Loftus, S.; Liu, H.; Bonde, G.; Sompallae, R.; Gildea, D.E.; Santana, J.F.; Manak, J.; Pavan, W.; et al. TFAP2 paralogs regulate melanocyte differentiation in parallel with MITF. PLoS Genet. 2017, 13, e1006636. [Google Scholar] [CrossRef] [PubMed]
- Kenny, C.; Dilshat, R.; Seberg, H.; Van Otterloo, E.; Bonde, G.; Helverson, A.; Steingrimsson, E.; Cornell, R.A. TFAP2 paralogs pioneer chromatin access for MITF and directly inhibit genes associated with cell migration. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hallberg, A.R.; Vorrink, S.U.; Hudachek, D.R.; Cramer-Morales, K.; Milhem, M.M.; Cornell, R.A.; Domann, F.E. Aberrant CpG methylation of the TFAP2A gene constitutes a mechanism for loss of TFAP2A expression in human metastatic melanoma. Epigenetics 2014, 9, 1641–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Wang, Q.-E.; Ray, A.; Wani, G.; Han, C.; Milum, K.; Wani, A.A. Modulation of Nucleotide Excision Repair by Mammalian SWI/SNF Chromatin-remodeling Complex. J. Biol. Chem. 2009, 284, 30424–30432. [Google Scholar] [CrossRef] [Green Version]
- Habel, N.; El-Hachem, N.; Soysouvanh, F.; Hadhiri-Bzioueche, H.; Giuliano, S.; Nguyen, S.; Horák, P.; Gay, A.-S.; Debayle, D.; Nottet, N.; et al. FBXO32 links ubiquitination to epigenetic reprograming of melanoma cells. Cell Death Differ. 2021, 28, 1837–1848. [Google Scholar] [CrossRef]
- Ondrušová, L.; Vachtenheim, J.; Réda, J.; Žáková, P.; Benková, K. MITF-Independent Pro-Survival Role of BRG1-Containing SWI/SNF Complex in Melanoma Cells. PLoS ONE 2013, 8, e54110. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Rao, Y.; Sun, Q.; Liu, Y.; Chen, J.; Bu, W. Long noncoding RNA CPS1-IT1 suppresses melanoma cell metastasis through inhibiting Cyr61 via competitively binding to BRG1. J. Cell. Physiol. 2019, 234, 22017–22027. [Google Scholar] [CrossRef] [PubMed]
- Laurette, P.; Coassolo, S.; Davidson, G.; Michel, I.; Gambi, G.; Yao, W.; Sohier, P.; Li, M.; Mengus, G.; LaRue, L.; et al. Chromatin remodellers Brg1 and Bptf are required for normal gene expression and progression of oncogenic Braf-driven mouse melanoma. Cell Death Differ. 2020, 27, 29–43. [Google Scholar] [CrossRef]
- Inoue, D.; Chew, G.-L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.W.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Reyes, J.C.; Barra, J.; Muchardt, C.; Camus, A.; Babinet, C.; Yaniv, M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α). EMBO J. 1998, 17, 6979–6991. [Google Scholar] [CrossRef]
- Martin, A.R.; Lin, M.; Granka, J.M.; Myrick, J.W.; Liu, X.; Sockell, A.; Atkinson, E.; Werely, C.J.; Möller, M.; Sandhu, M.S.; et al. An Unexpectedly Complex Architecture for Skin Pigmentation in Africans. Cell 2017, 171, 1340–1353.e14. [Google Scholar] [CrossRef] [Green Version]
- Moloney, F.J.; Lyons, G.; Bock, V.L.; Huang, X.X.; Bugeja, M.J.; Halliday, G.M. Hotspot Mutation of Brahma in Non-Melanoma Skin Cancer. J. Investig. Dermatol. 2009, 129, 1012–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, D.; Curry, J.L.; Lin, Q.; Richards, H.W.; Chen, D.; Hornsby, P.J.; Timchenko, N.A.; Medrano, E.E. Dynamic assembly of chromatin complexes during cellular senescence: Implications for the growth arrest of human melanocytic nevi. Aging Cell 2007, 6, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaros, S.; Cirrincione, G.M.; Muchardt, C.; Kleer, C.G.; Michael, C.W.; Reisman, D. The reversible epigenetic silencing of BRM: Implications for clinical targeted therapy. Oncogene 2007, 26, 7058–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchardt, C.; Bourachot, B.; Reyes, J.; Yaniv, M. ras transformation is associated with decreased expression of the brm/SNF2alpha ATPase from the mammalian SWI-SNF complex. EMBO J. 1998, 17, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Saladi, S.V.; Trivedi, A.R.; Aras, S.; Qi, H.; Jayanthy, A.; Setaluri, V.; de la Serna, I.L. Modulation of BRAHMA expression by the mitogen-activated protein kinase/extracellular signal regulated kinase pathway is associated with changes in melanoma proliferation. Arch. Biochem. Biophys. 2014, 563, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [Green Version]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, R.; Chanana, U.B.; Hussain, S.; Sharma, S.; Goel, K.; Bisht, D.; Patne, K.; Swer, P.B.; Hockensmith, J.W.; Muthuswami, R. Altering mammalian transcription networking with ADAADi: An inhibitor of ATP-dependent chromatin remodeling. PLoS ONE 2021, 16, e0251354. [Google Scholar] [CrossRef] [PubMed]
- Rago, F.; Rodrigues, L.U.; Bonney, M.; Sprouffske, K.; Kurth, E.; Elliott, G.; Ambrose, J.; Aspesi, P.; Oborski, J.; Chen, J.T.; et al. Exquisite Sensitivity to Dual BRG1/BRM ATPase Inhibitors Reveals Broad SWI/SNF Dependencies in Acute Myeloid Leukemia. Mol. Cancer Res. 2022, 20, 361–372. [Google Scholar] [CrossRef]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Lee, D.Y.; Hwang, Y.S.; Seo, H.R.; Lee, S.A.; Kwon, J. The Bromodomain Inhibitor PFI-3 Sensitizes Cancer Cells to DNA Damage by Targeting SWI/SNF. Mol. Cancer Res. 2021, 19, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Y.; Sims, M.M.; He, Y.; Miller, D.D.; Pfeffer, L.M. Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma. Pharmaceuticals 2021, 14, 904. [Google Scholar] [CrossRef] [PubMed]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; Mueller, W.; Weber, M.; Sévenet, N.; Delattre, O.; von Deimling, A. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br. J. Cancer 2001, 84, 199–201. [Google Scholar] [CrossRef]
- Hadfield, K.D.; Newman, W.G.; Bowers, N.L.; Wallace, A.; Bolger, C.; Colley, A.; McCann, E.; Trump, D.; Prescott, T.; Evans, D.G.R. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008, 45, 332–339. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Collings, C.K.; Dao, H.T.; Pierre, R.S.; Cheng, Y.-C.; Huang, J.; Sun, Z.-Y.; Seo, H.-S.; Mashtalir, N.; Comstock, D.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e23. [Google Scholar] [CrossRef] [PubMed]
- Guidi, C.J.; Sands, A.T.; Zambrowicz, B.P.; Turner, T.K.; Demers, D.A.; Webster, W.; Smith, T.W.; Imbalzano, A.N.; Jones, S.N. Disruption of Ini1 Leads to Peri-Implantation Lethality and Tumorigenesis in Mice. Mol. Cell. Biol. 2001, 21, 3598–3603. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.; Galusha, S.A.; McMenamin, M.E.; Fletcher, C.D.M.; Orkin, S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 13796–13800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; Mir, S.N.; Wani, G.; Zhao, Q.; Battu, A.; Zhu, Q.; Wang, Q.-E.; Wani, A.A. Human SNF5/INI1, a Component of the Human SWI/SNF Chromatin Remodeling Complex, Promotes Nucleotide Excision Repair by Influencing ATM Recruitment and Downstream H2AX Phosphorylation. Mol. Cell. Biol. 2009, 29, 6206–6219. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Wong, R.P.C.; Martinka, M.; Li, G. Loss of SNF5 Expression Correlates with Poor Patient Survival in Melanoma. Clin. Cancer Res. 2009, 15, 6404–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scurr, L.L.; Pupo, G.M.; Becker, T.M.; Lai, K.; Schrama, D.; Haferkamp, S.; Irvine, M.; Scolyer, R.A.; Mann, G.J.; Becker, J.C.; et al. IGFBP7 Is Not Required for B-RAF-Induced Melanocyte Senescence. Cell 2010, 141, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; de Oliveira, R.L.; Wang, C.; Neto, J.M.F.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alajem, A.; Biran, A.; Harikumar, A.; Sailaja, B.S.; Aaronson, Y.; Livyatan, I.; Nissim-Rafinia, M.; Sommer, A.G.; Mostoslavsky, G.; Gerbasi, V.R.; et al. Differential Association of Chromatin Proteins Identifies BAF60a/SMARCD1 as a Regulator of Embryonic Stem Cell Differentiation. Cell Rep. 2015, 10, 2019–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Jothi, R.; Ronan, J.L.; Cui, K.; Zhao, K.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. USA 2009, 106, 5187–5191. [Google Scholar] [CrossRef] [Green Version]
- Witzel, M.; Petersheim, D.; Fan, Y.; Bahrami, E.; Racek, T.; Rohlfs, M.; Puchałka, J.; Mertes, C.; Gagneur, J.; Ziegenhain, C.; et al. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat. Genet. 2017, 49, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; Kadoch, C. A SMARCD2-containing mSWI/SNF complex is required for granulopoiesis. Nat. Genet. 2017, 49, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Zhu, L.; Gao, Y.; Zhang, X.; Yan, Y.; Cen, J.; Li, R.; Zeng, R.; Liao, L.; Hou, C.; et al. Baf60b-mediated ATM-p53 activation blocks cell identity conversion by sensing chromatin opening. Cell Res. 2017, 27, 642–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Saladi, S.V.; Basuroy, T.; Marathe, H.G.; Lorès, P.; de la Serna, I.L. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation. J. Cell. Physiol. 2019, 234, 11780–11791. [Google Scholar] [CrossRef] [PubMed]
- Weider, M.; Küspert, M.; Bischof, M.; Vogl, M.R.; Hornig, J.; Loy, K.; Kosian, T.; Müller, J.; Hillgärtner, S.; Tamm, E.; et al. Chromatin-Remodeling Factor Brg1 Is Required for Schwann Cell Differentiation and Myelination. Dev. Cell 2012, 23, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, C.; Forcales, S.; Hill, D.A.; Imbalzano, A.N.; Latella, L.; Puri, P.L. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004, 36, 738–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.-X.; Li, S.; Wang, L.; Ko, H.J.; Lee, Y.; Jung, D.Y.; Okutsu, M.; Yan, Z.; Kim, J.K.; Lin, J.D. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nat. Med. 2013, 19, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wong, R.H.; Tang, T.; Hudak, C.S.; Yang, D.; Duncan, R.E.; Sul, H.S. Phosphorylation and Recruitment of BAF60c in Chromatin Remodeling for Lipogenesis in Response to Insulin. Mol. Cell 2012, 49, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.-D.; Zhao, X.; Li, W.-T. Identification of differentially expressed metastatic genes and their signatures to predict the overall survival of uveal melanoma patients by bioinformatics analysis. Int. J. Ophthalmol. 2020, 13, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer gene mutation frequencies for the U.S. population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [PubMed]
- Ticha, I.; Hojny, J.; Michalkova, R.; Kodet, O.; Krkavcova, E.; Hajkova, N.; Nemejcova, K.; Bartu, M.; Jaksa, R.; Dura, M.; et al. A comprehensive evaluation of pathogenic mutations in primary cutaneous melanomas, including the identification of novel loss-of-function variants. Sci. Rep. 2019, 9, 17050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Joseph, N.M.; Yu, R.; Benhamida, J.; Liu, S.; Prow, T.; Ruben, B.; North, J.; Pincus, L.; Yeh, I.; et al. Abstract NG07: Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Bioinform. Syst. Biol. 2018, 78, NG07. [Google Scholar] [CrossRef]
- Váraljai, R.; Horn, S.; Sucker, A.; Piercianek, D.; Schmitt, V.; Carpinteiro, A.; Becker, K.; Reifenberger, J.; Roesch, A.; Felsberg, J.; et al. Integrative Genomic Analyses of Patient-Matched Intracranial and Extracranial Metastases Reveal a Novel Brain-Specific Landscape of Genetic Variants in Driver Genes of Malignant Melanoma. Cancers 2021, 13, 731. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W.; Zhang, Y.; Cieślik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Investig. 2020, 130, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Wu, S.; Fukumoto, T.; Lin, J.; Nacarelli, T.; Wang, Y.; Ong, D.; Liu, H.; Fatkhutdinov, N.; Zundell, J.A.; Karakashev, S.; et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat. Cancer 2021, 2, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.-M.; Conejo-Garcia, J.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.C.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Meersma, G.J.; Lieftink, C.; Beijersbergen, R.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.-F.; Cao, J.; Burley, S.K.; Wang, H.-Y.; Zheng, X.S. mTORC1 Promotes ARID1A Degradation and Oncogenic Chromatin Remodeling in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 5652–5665. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Probst, L.; Wain, H.M.; Maltais, L.; Tucker, P.W.; Moran, E. Nomenclature of the ARID family of DNA-binding proteins. Genomics 2005, 86, 242–251. [Google Scholar] [CrossRef]
- Lee, J.J.; Sholl, L.M.; Lindeman, N.I.; Granter, S.R.; Laga, A.C.; Shivdasani, P.; Chin, G.; Luke, J.J.; Ott, P.A.; Hodi, F.S.; et al. Targeted next-generation sequencing reveals high frequency of mutations in epigenetic regulators across treatment-naïve patient melanomas. Clin. Epigenetics 2015, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Broit, N.; Johansson, P.A.; Rodgers, C.B.; Walpole, S.T.; Newell, F.; Hayward, N.K.; Pritchard, A.L. Meta-Analysis and Systematic Review of the Genomics of Mucosal Melanoma. Mol. Cancer Res. 2021, 19, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, R.; Ma, X.; Judson-Torres, R.L.; Zeng, H. Mucosal Melanoma: Pathological Evolution, Pathway Dependency and Targeted Therapy. Front. Oncol. 2021, 11, 702287. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, K.; Jia, Y.; Chuang, J.-C.; Sun, X.; Lin, Y.-H.; Celen, C.; Li, L.; Huang, F.; Liu, X.; et al. Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nat. Cancer 2020, 1, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, M.; Kanchi, K.L.; Dees, N.D.; Lu, C.; Griffith, M.; Fenstermacher, D.; Sung, H.; Miller, C.A.; Goetz, B.; et al. Clonal Architectures and Driver Mutations in Metastatic Melanomas. PLoS ONE 2014, 9, e111153. [Google Scholar] [CrossRef] [PubMed]
- Hodges, H.C.; Kirkland, J.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, T.; Monterde, B.; González-Silva, L.; Betancor-Fernández, I.; Revilla, C.; Agraz-Doblas, A.; Freire, J.; Isidro, P.; Quevedo, L.; Blanco, R.; et al. ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to chemotherapy in lung cancer. Oncogene 2021, 40, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Künzel, J.; Löbrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in Transcriptional Repression and Repair at DNA Breaks in Actively Transcribed Regions of Chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Fewings, E.; Chang, D.; Zeng, H.; Liu, S.; Jorapur, A.; Belote, R.L.; McNeal, A.S.; Tan, T.M.; Yeh, I.; et al. The genomic landscapes of individual melanocytes from human skin. Nature 2020, 586, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, T.; Lin, J.; Fatkhutdinov, N.; Liu, P.; Somasundaram, R.; Herlyn, M.; Zhang, R.; Nishigori, C. ARID2 Deficiency Correlates with the Response to Immune Checkpoint Blockade in Melanoma. J. Investig. Dermatol. 2021, 141, 1564–1572.e4. [Google Scholar] [CrossRef]
- Jiang, H.; Cao, H.-J.; Ma, N.; Bao, W.-D.; Wang, J.-J.; Chen, T.-W.; Zhang, E.-B.; Yuan, Y.-M.; Ni, Q.-Z.; Zhang, F.-K.; et al. Chromatin remodeling factor ARID2 suppresses hepatocellular carcinoma metastasis via DNMT1-Snail axis. Proc. Natl. Acad. Sci. USA 2020, 117, 4770–4780. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; de Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.-L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Cui, K.; Murray, D.M.; Ling, C.; Xue, Y.; Gerstein, A.; Parsons, R.; Zhao, K.; Wang, W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhai, W.; Richardson, J.A.; Olson, E.N.; Meneses, J.J.; Firpo, M.T.; Kang, C.; Skarnes, W.C.; Tjian, R. Polybromo protein BAF180 functions in mammalian cardiac chamber maturation. Genes Dev. 2004, 18, 3106–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanetzki, V.; Fröb, F.; Baroti, T.; Schimmel, M.; Tamm, E.R.; Wegner, M. Role of the Pbrm1 subunit and the PBAF complex in Schwann cell development. Sci. Rep. 2022, 12, 2651. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef]
- Talantov, D.; Mazumder, A.; Yuqiu, J.; Briggs, T.; Jiang, Y.; Backus, J.; Atkins, D.; Wang, Y. Novel Genes Associated with Malignant Melanoma but not Benign Melanocytic Lesions. Clin. Cancer Res. 2005, 11, 7234–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Shen, S.; Hoshida, Y.; Subramanian, A.; Ross, K.; Brunet, J.-P.; Wagner, S.; Ramaswamy, S.; Mesirov, J.P.; Hynes, R.O. Gene Expression Changes in an Animal Melanoma Model Correlate with Aggressiveness of Human Melanoma Metastases. Mol. Cancer Res. 2008, 6, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Mason, L.D.; Chava, S.; Reddi, K.K.; Gupta, R. The BRD9/7 Inhibitor TP-472 Blocks Melanoma Tumor Growth by Suppressing ECM-Mediated Oncogenic Signaling and Inducing Apoptosis. Cancers 2021, 13, 5516. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liu, Y.; Wu, C.; Li, M.; Wei, Y.; Niu, W.; Yang, J.; Fan, S.; Xie, Y.; Li, H.; et al. BRD7 Promotes Cell Proliferation and Tumor Growth through Stabilization of c-Myc in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 659392. [Google Scholar] [CrossRef]
- Yamamoto, J.; Suwa, T.; Murase, Y.; Tateno, S.; Mizutome, H.; Asatsuma-Okumura, T.; Shimizu, N.; Kishi, T.; Momose, S.; Kizaki, M.; et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat. Chem. Biol. 2020, 16, 1208–1217. [Google Scholar] [CrossRef]
- Vorobyeva, N.E.; Nikolenko, J.V.; Nabirochkina, E.N.; Krasnov, A.N.; Shidlovskii, Y.V.; Georgieva, S. SAYP and Brahma are important for ‘repressive’ and ‘transient’ Pol II pausing. Nucleic. Acids Res. 2012, 40, 7319–7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaka, A.; Mizutani, T.; Kobayashi, K.; Tando, T.; Sakurai, K.; Fujiwara, T.; Iba, H. Double plant homeodomain (PHD) finger proteins DPF3a and -3b are required as transcriptional co-activators in SWI/SNF complex-dependent activation of NF-kappaB RelA/p50 heterodimer. J. Biol. Chem. 2012, 287, 11924–11933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbunathan, H.; Verstraten, R.; Singh, A.D.; Harbour, J.W.; Bowcock, A.M. Integrative Copy Number Analysis of Uveal Melanoma Reveals Novel Candidate Genes Involved in Tumorigenesis Including a Tumor Suppressor Role for PHF10/BAF45a. Clin. Cancer Res. 2019, 25, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Soshnikova, N.V.; Tatarskiy, E.V.; Klimenko, N.S.; Shtil, A.A.; Nikiforov, M.A.; Georgieva, S.G. PHF10 subunit of PBAF complex mediates transcriptional activation by MYC. Oncogene 2021, 40, 6071–6080. [Google Scholar] [CrossRef]
- Wei, M.; Liu, B.; Su, L.; Li, J.; Zhang, J.; Yu, Y.; Yan, M.; Yang, Z.; Chen, X.; Liu, J.; et al. A Novel Plant Homeodomain Finger 10–Mediated Antiapoptotic Mechanism Involving Repression of Caspase-3 in Gastric Cancer Cells. Mol. Cancer Ther. 2010, 9, 1764–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Nie, H.; Wang, M.; Su, L.; Li, J.; Yu, B.; Wei, M.; Ju, J.; Yu, Y.; Yan, M.; et al. MicroRNA-409-3p regulates cell proliferation and apoptosis by targeting PHF10 in gastric cancer. Cancer Lett. 2012, 320, 189–197. [Google Scholar] [CrossRef]
- Sinha, S.; Biswas, M.; Chatterjee, S.S.; Kumar, S.; Sengupta, A. Pbrm1 Steers Mesenchymal Stromal Cell Osteolineage Differentiation by Integrating PBAF-Dependent Chromatin Remodeling and BMP/TGF-beta Signaling. Cell Rep. 2020, 31, 107570. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, B.C.; D’Avino, A.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.; Valencia, A.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, J.; Jiang, J.; Yuan, J.; Zhang, Y.; Wei, X.; Loo, N.; Wang, Y.; Pan, Y.; Zhang, T.; et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 2019, 10, 733. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Sun, X.; Zhao, Z.; Sun, H.; Sun, P. BRD9 inhibition promotes PUMA-dependent apoptosis and augments the effect of imatinib in gastrointestinal stromal tumors. Cell Death Dis. 2021, 12, 962. [Google Scholar] [CrossRef] [PubMed]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Debbache, J.; Peña-Hernández, R.; Antunes, A.T.; Schaefer, S.M.; Cheng, P.; Zimmerli, D.; Haeusel, J.; Calçada, R.R.; Tuncer, E.; et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell 2018, 34, 69–84.e14. [Google Scholar] [CrossRef]
- Alver, B.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W.M. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef]
- Baggiolini, A.; Callahan, S.J.; Montal, E.; Weiss, J.M.; Trieu, T.; Tagore, M.M.; Tischfield, S.E.; Walsh, R.M.; Suresh, S.; Fan, Y.; et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science 2021, 373, 1048. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Nittner, D.; Rambow, F.; Radaelli, E.; Stanchi, F.; Vandamme, N.; Baggiolini, A.; Sommer, L.; Berx, G.; Oord, J.J.V.D.; et al. Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem. Cell 2017, 21, 679–693.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, R.S.; Alver, B.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef] [PubMed]



| Gene | Total | Truncating Mutation | Deep Deletion | Splice Mutation | Missense Mutation | In frame Mutation | Structural Variant | Amplification |
|---|---|---|---|---|---|---|---|---|
| ARID2 | 18% | 41% | 0 | 5% | 47% | 0 | 1% | 8% |
| (78) | (32) * | (4) | (37) * | (1) | (6) ** | |||
| SMARCA4 | 10% | 7% | 0 | 4% | 89% | 0 | 0 | 0 |
| (46) | (3) | (2) | (41) | |||||
| ARID1A | 9% | 22% | 2% | 10% | 59% | 0 | 5% | 2% |
| (41) | (9) | (1) | (4) | (24) | (2) | (1) * | ||
| SMARCA2 | 9% | 15% | 5% | 0 | 74% | 0 | 5% | 3% |
| (39) | (6) | (2) | (29) | (2) | (1) | |||
| ARID1B | 9% | 26% | 15% | 0 | 56% | 0 | 3% | 0 |
| (39) | (10) | (6) | (22) | (1) | ||||
| BRD9 | 8% | 3% | 5% | 5% | 41% | 0 | 3% | 46% |
| (37) | (1) * | (2) | (2) | (15) | (1) | (17) * | ||
| PBRM1 | 8% | 36% | 0 | 6% | 61% | 0 | 6% | 3% |
| (36) | (9) | (2) | (22) | (2) | (1) | |||
| SMARCC2 | 8% | 21% | 0 | 6% | 65% | 3% | 0 | 6% |
| (34) | (7) | (2) | (22) | (1) | (2) | |||
| BICRAL | 8% | 3% | 0 | 0 | 59% | 0 | 0 | 38% |
| (34) | (1) | (20) | (13) | |||||
| SMARCC1 | 6% | 7% | 0 | 0 | 89% | 0 | 0 | 4% |
| (27) | (2) | (24) | (1) | |||||
| ACTL6B | 6% | 4% | 0 | 8% | 76% | 0 | 0 | 12% |
| (25) | (1) | (2) | (19) | (3) | ||||
| BRD7 | 5% | 41% | 14% | 5% | 41% | 0 | 0 | 0 |
| (22) | (9) | (3) | (1) | (9) | ||||
| DPF2 | 5% | 0 | 14% | 5% | 52% | 0 | 5% | 24% |
| (21) | (3) | (1) | (11) | (1) | (5) | |||
| ACTB | 5% | 5% | 0 | 5% | 43% | 0 | 0 | 48% |
| (21) | (1) | (1) | (9) | (10) | ||||
| SMARCD3 | 4% | 6% | 6% | 0 | 44% | 0 | 0 | 44% |
| (18) | (1) | (1) | (8) | (8) | ||||
| SMARCD2 | 3% | 0 | 0 | 7% | 40% | 0 | 7% | 53% |
| (15) | (1) | (6) | (1) * | (8) * | ||||
| PHF10 | 3% | 7% | 36% | 0 | 57% | 0 | 0 | 0 |
| (14) | (1) | (5) | (8) | |||||
| SMARCD1 | 3% | 0 | 0 | 15% | 53% | 0 | 0 | 23% |
| (13) | (2) | (8) | (3) | |||||
| DPF3 | 3% | 0 | 15% | 8% | 77% | 0 | 0 | 0 |
| (13) | (2) | (1) | 10 | |||||
| BCL7C | 3% | 8% | 0 | 0 | 83% | 0 | 0 | 8% |
| (12) | (1) | (10) | (1) | |||||
| BICRA | 2% | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| (10) | (10) | |||||||
| SMARCB1 | 2% | 10% | 10% | 0 | 50% | 0 | 0 | 30% |
| (10) | (1) | (1) | (5) | (3) | ||||
| ACTL6A | 2% | 10% | 20% | 10% | 40% | 0 | 0 | 20% |
| (10) | (1) | (2) | (1) | (4) | (2) | |||
| DPF1 | 2% | 0 | 11% | 0 | 67% | 0 | 0 | 22% |
| (9) | (1) | (6) | (2) | |||||
| SS18 | 2% | 11% | 0 | 11% | 78% | 0 | 0 | 0 |
| (9) | (1) | (1) | (7) | |||||
| SMARCE1 | 2% | 13% | 0 | 13% | 63% | 0 | 0 | 13% |
| (8) | (1) | (1) | (5) | (1) | ||||
| BCL7B | 2% | 25% | 0 | 0 | 50% | 0 | 0 | 25% |
| (8) | (2) | (4) | (2) | |||||
| BCL7A | 1% | 0 | 0 | 17% | 50% | 0 | 0 | 33% |
| (6) | (1) | (3) | (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreier, M.R.; de la Serna, I.L. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes 2022, 6, 10. https://doi.org/10.3390/epigenomes6010010
Dreier MR, de la Serna IL. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes. 2022; 6(1):10. https://doi.org/10.3390/epigenomes6010010
Chicago/Turabian StyleDreier, Megan R., and Ivana L. de la Serna. 2022. "SWI/SNF Chromatin Remodeling Enzymes in Melanoma" Epigenomes 6, no. 1: 10. https://doi.org/10.3390/epigenomes6010010
APA StyleDreier, M. R., & de la Serna, I. L. (2022). SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes, 6(1), 10. https://doi.org/10.3390/epigenomes6010010