1. Introduction
Aside from post-translational histone modifications and small RNA populations, the epigenome of an organism is defined by the level and spectrum of DNA methylation [
1]. Cytosine methylation can occur as 5-methylcytosine (5mC) or 5-hydroxymethylcytosine (5hmC) [
2]. Its key roles are in embryonic development regulation, genome imprinting, silencing of transposons, cell differentiation, X-chromosome inactivation, and general transcriptional gene regulation [
3,
4]. The addition of methyl groups onto DNA bases generally represses gene expression and therefore acts as one of several transcription control mechanisms.
Whole genome bisulfite-sequencing has become the gold standard to detect DNA methylation patterns because of its single-base resolution and the possibility to cover entire genomes. Other approaches use either pre-selection and single-base resolution [
5] or region-based approaches in whole genomes [
6,
7] and, therefore, do not deliver the full spectrum and detail of DNA methylation patterns.
Since the development of bisulfite-treated DNA sequencing in 1992 by Frommer et al. [
8], the application of high-throughput sequencing has been especially useful in facilitating the reliable detection and analysis of methylation patterns in several organisms, tissues, and cell types. Huge steps in the field of DNA methylation analysis were the first WGBS of
Arabidopsis thaliana [
9,
10], and the first human methylome sequencing in 2009 by Lister et al. [
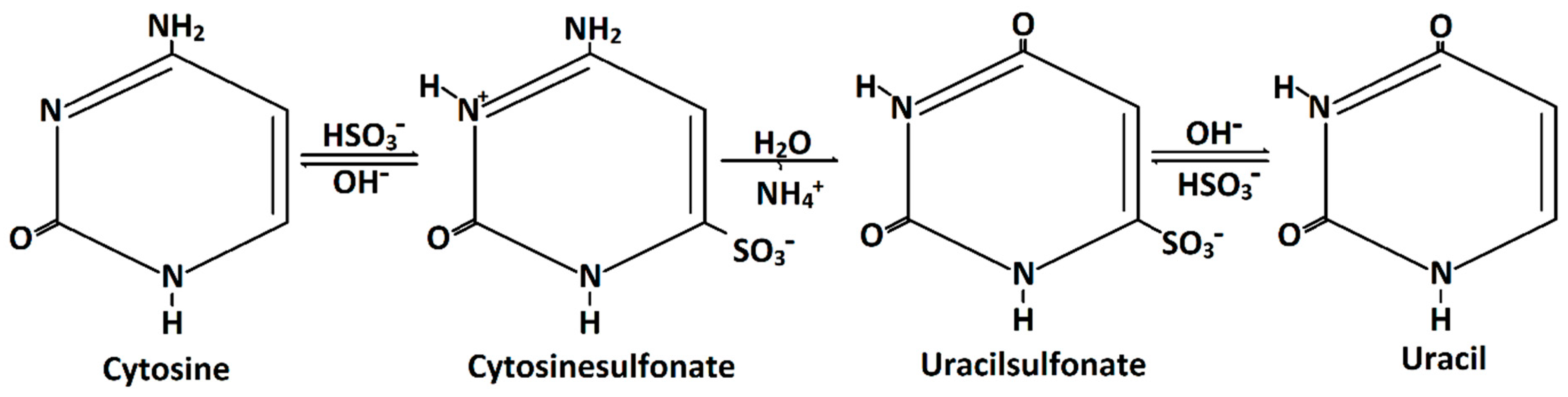
11]. During the bisulfite reaction of DNA, unmethylated cytosine is converted to uracil (
Figure 1). This occurrs via cytosinsulfonate, a further hydrolytic deamination step to uracilsulfonate, and, finally, a desulfonation step to uracil (
Figure 1). However, 5mC and 5hmC are inert to this chemical conversion.
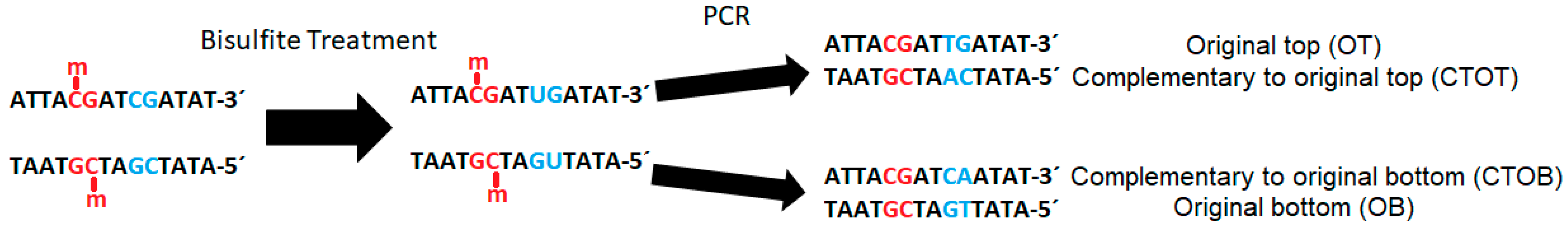
The subsequent polymerase chain reaction (PCR) translates uracil into thymine. During sequencing, this base pair shift causes cytosine/thymine-polymorphism, which can be quantified and interpreted as proportion of the original methylation at a specific site through the comparison of reads with the original strand or a reference genome (
Figure 2).
A disadvantage of WGBS is that bisulfite sequencing cannot distinguish between 5hmC and 5mC. The 5hmC is described as the dynamic DNA modification associated with the DNA demethylation process [
12] in animals. This has to be taken into account for organisms/cell types with substantial amounts of 5hmC, such as the cerebellum cells of Parkinson disease patients [
13]. In plants, 5hmC is of minor relevance [
14], as it occurs only in very low amounts and may merely serve as an intermediate product during demethylation [
15].
A second disadvantage arises from the C–T base switch, particularly when analyzing non-reference strains or mutagenesis populations, as it can become impossible to distinguish between bisulfite-induced deamination of unmethylated cytosine on the one hand and true C-to-T single nucleotide polymorphism (SNP) on the other.
In plants and some animals, large parts of the genome are methylated depending on the genome size, genome duplications, and the control of most of these duplicated parts. In most animals, cytosine carries methylation predominantly in the CG context accumulated in CpG islands for gene regulation, often near transcription starting sites [
16]. In contrast to that, plant genomes have additional CHG and CHH methylation (H = adenine, thymine or cytosine). These last two correlate with the suppression of transposon activity [
17], mobile genetic elements that can change their position within the genome. While CG and CHG methylation are symmetric, i.e., the palindromic cytosine on the complementary strand is usually also methylated, CHH methylation is asymmetrical [
18]. The proportion and context of cytosine methylation ranges from <1% total cytosine methylation during the early development of
Drosophila melanogaster [
19] to 74.7% CG, 69.1% CHG, and 1.5% CHH methylation in
Picea abies [
20].
DNA methylation patterns are often tissue-specific [
21], environment-altered [
22,
23], and can be stably inherited to subsequent generations [
24,
25].
Because of the importance of DNA methylation as transcriptional regulator, its analysis plays an increasing role in integrative -omics studies, ecological examinations, and medical research. The aim of this publication is to facilitate the launch of WGBS experiments. Therefore, we will give a brief overview of WGBS experiment planning by discussing different types of WGBS library preparation protocols, options for sequencing technology, and bioinformatics data analysis.
2. Whole Genome Bisulfite-Sequencing Preparation
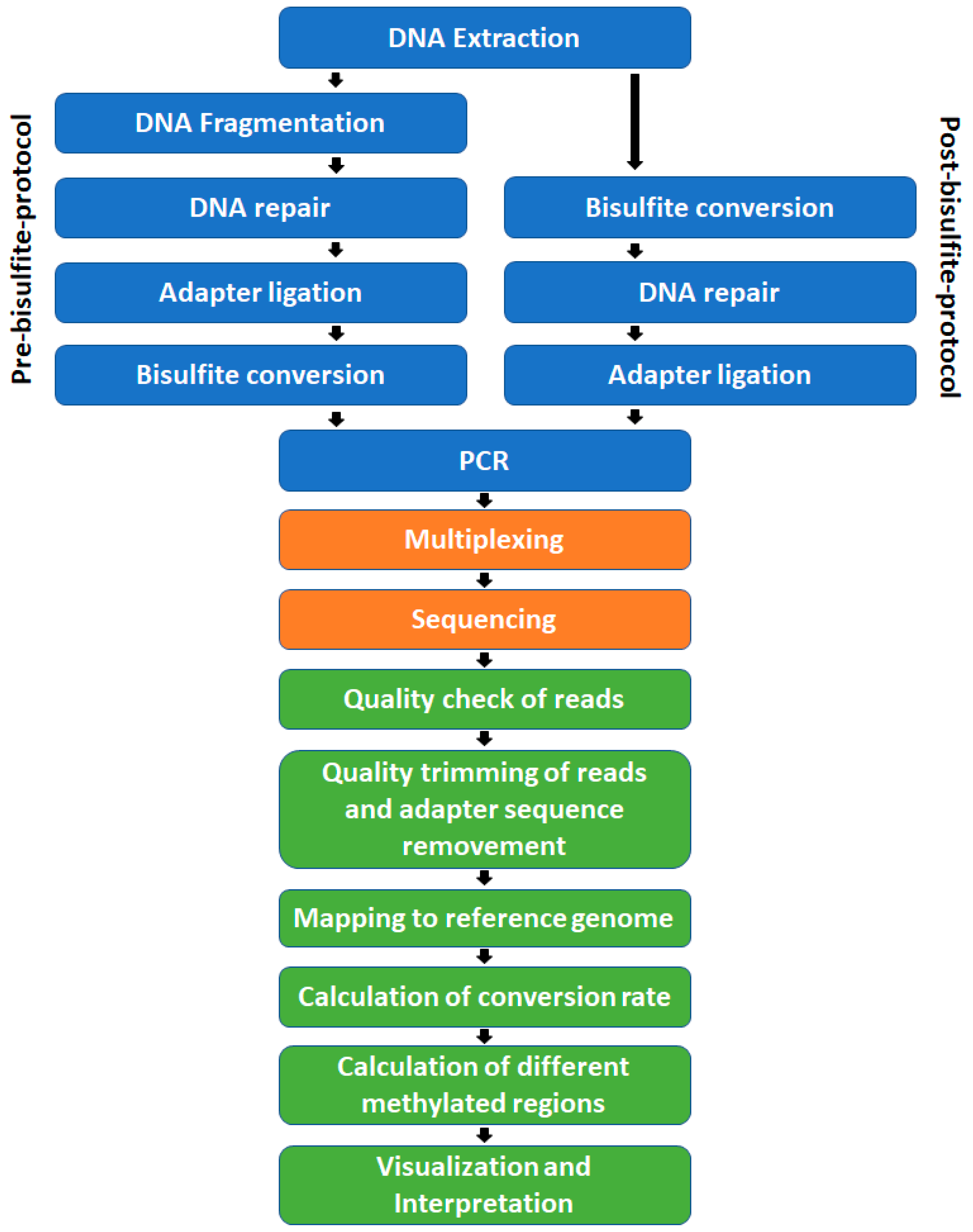
The steps, prior to sequencing, needed for WGBS library preparation are: DNA extraction, DNA fragmentation, DNA repair, adapter ligation, bisulfite treatment, PCR amplification (
Figure 3) including size selection, and quality control at different points in time. These can be arranged in two general ways. The pathway shown on the left side of
Figure 3 is a pre-bisulfite protocol. Because the bisulfite treatment takes place after the adapter ligation, these protocols require methylated adapters that will be inert to the treatment. In post-bisulfite protocols, like a post-bisulfite adapter tagging protocol, DNA fragmentation is facilitated through the bisulfite treatment itself and is followed by DNA end repair and adapter ligation or random priming including streptavidin-coated magnetic beads [
26]. The advantage of the latter is that less DNA is needed as less adapter-ligated DNA fragments are destroyed by the harsh conditions of bisulfite treatment [
27]. A disadvantage of this kind of library preparation is the slightly higher computational power needed to bioinformatically process these non-directional reads. Usually, over 90% of the adapter-ligated, intact fragments are destroyed in pre-bisulfite protocols, even under ideal conditions [
28]. In the end, the bisulfite conversion step leads to the conversion of cytosine to uracil, which is much more susceptible to degradation [
29]. The decline in the amount of DNA could be compensated by PCR amplification, but should be done with as few cycles as possible to yield the lowest possible PCR bias. Minimum two PCR cycles are needed because the former unmethylated cytosines have to be rewritten from uracil to thymine. The used polymerase must have the ability to read uracil. Therefore, most polymerases with proofreading and repair functions, except of Pfu Turbo Cx, cannot be used. Another common polymerase for this purpose is KAPA HiFi Uracil+. Furthermore, highly PCR-amplified bisulfite-converted libraries may be unbalanced, as highly methylated, and fragments with high C content post-conversion are easier to amplify and, therefore, will be overrepresented in the final library [
30].
The fragment length depends on the sequencing technology. The desired read length could be adjusted after DNA shearing by gel selection, bead selection, or BluePippin (Sage Science), an automated DNA and RNA size selection system. Depending on the final sequencing platform, fragments should have a length of 200–400 base pairs (without adapters; see Sequencing technology). Classic gel selection has to be made, for example, with SYBR Gold stain instead of ethidium bromide to avoid DNA degradation and contamination. The desired fragment length could be easily cut out of a 1% agarose gel after DNA fragmentation if a DNA standard has also been run on the same gel. This process was automated in BluePippin by the application of a gel cassette. However, most library preparation kits today use magnetic beads for size selection.
To calculate the bisulfite conversion rate, unmethylated reference DNA has to be added to the library prior to the bisulfite treatment, at a ratio of 0.1–0.5% (
w/
w) of the total DNA. Lambda phage DNA is most commonly used for this purpose. It is completely unmethylated and is, therefore, expected to be converted at every cytosine position. In plants with assembled chloroplast genomes, the unmethylated chloroplast DNA, which is contained to some degree in all genomic DNA extracts, can be used instead of or on top of lambda. In non-plant species it is also possible to use a non-CG methylation context to control the conversion efficiency [
31].
Several protocols for WGBS library preparation have been proposed using kits from different companies. The decision depends heavily on the experience of the individual laboratory and, therefore, is highly subjective. In general, a fast and routine working process is of importance to avoid batch effects and the introduction of bias due to amplification. This should be avoided by working with amplification-free libraries [
32,
33].
General examples for library preparation kits are EpiTect Plus (Qiagen, Venlo, the Netherlands), EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA), or Imprint® DNA Modification Kit from Sigma-Aldrich/Merck (St. Louis, MO, USA) for bisulfite conversion and Accel-NGS® Methyl-Seq DNA Library Kit (Swift Bioscience, Ann Arbor, MI, USA), TrueSeq Nano (Illumina, San Diego, CA, USA), or SureSelectXT Methyl-Seq Target Enrichment System (Agilent, Santa Clara, CA, USA) for adapter ligation.
The cost of a WGBS library is currently in the range of 50–240 €/sample (2018), depending on the library preparation method, supplier, and conversion kits.
3. Sequencing Technology
Aside from Roche, Thermo Fisher Scientific Inc., Beckman Coulter, and Pacific Biosciences, the market leader with more than 75% of the market share in terms of next generation sequencing technology is Illumina [
34], offering several platforms for different applications. For detailed information see the Illumina platform website (
https://emea.illumina.com/systems/sequencing-platforms.html).
Due to DNA degradation during the bisulfite treatment, the production of long fragments for sequencing is limited. The longer the fragment, the more likely is a fracture of the strand due to bisulfite treatment. Therefore, the advantages of long fragment generating systems, such as more reliable mapping of reads, currently do not apply. Benchtop solutions are partly not suitable because of the much higher price per giga base (Gb) of produced sequence. A comparison of the most promising systems currently used, such as HiSeq, HiSeqXTen, NovaSeq, and PacBio, is discussed in the following section.
For high-throughput sequencing, several systems exist, such as HiSeq2500, HiSeq3000 and HiSeq4000. They differ in terms of run time, maximum read length, and read output, but also in their acceptance of certain types of libraries, their potential to deal with low complexity libraries, and their quality score output. Compared to HiSeq3000 and HiSeq4000, the HiSeq2500 system can deal with low sequence diversity libraries like amplicon or WGBS libraries because of an optimization of the cluster calling algorithm. Nevertheless, the other systems could also be used if the libraries had been correctly multiplexed with other libraries with balanced base proportions at each read position or minimum 20% phi X DNA (see Multiplexing).
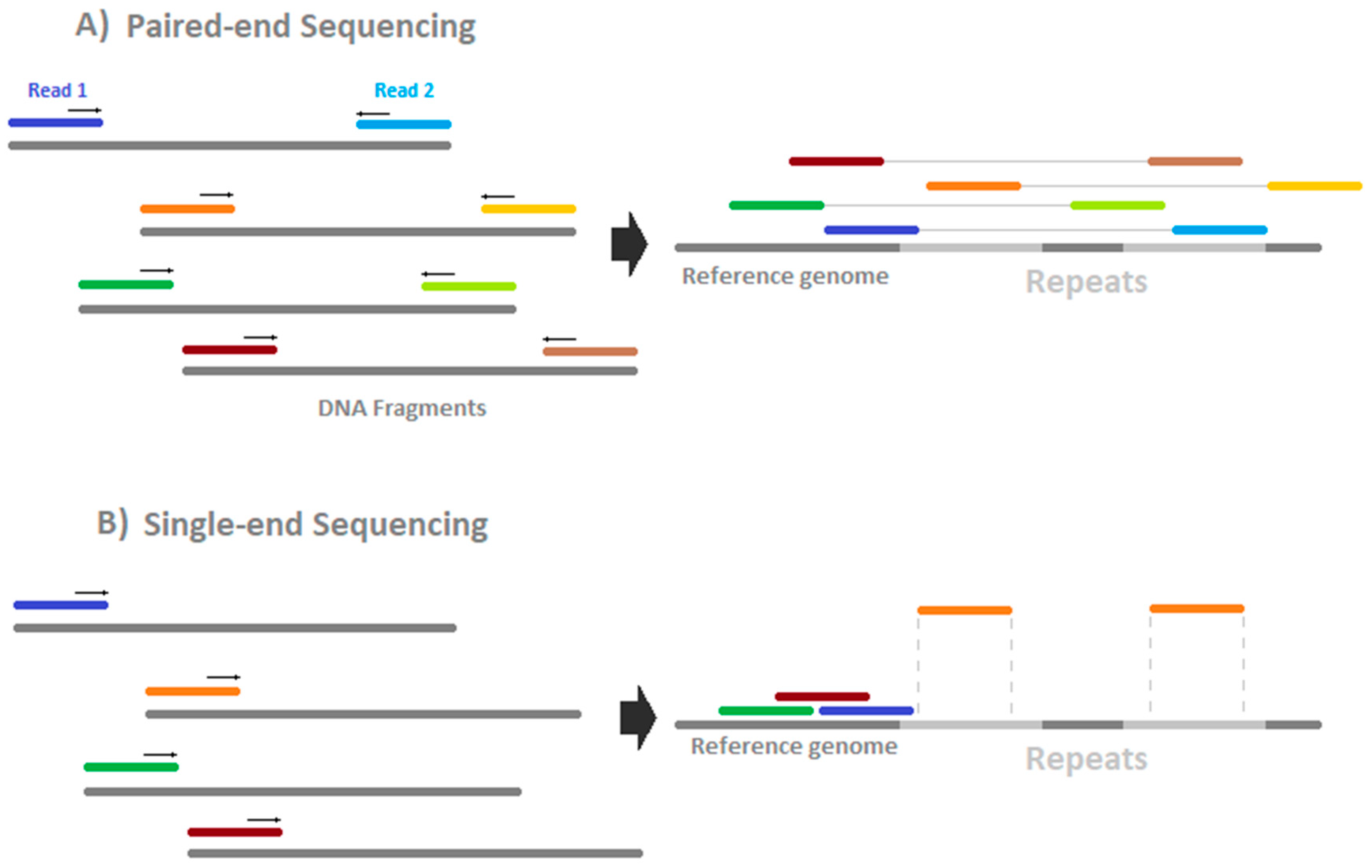
HiSeq sequencing costs are around 60–90 €/Gb (2018) depending on the read length and the option of paired- or single-end sequencing (see
Figure 4). Theoretically, paired-end sequencing offers the potential to map deeper into repeat-regions because two reads are generated, one from either side of the fragment. This has, so far, not been investigated systematically but seems to depend on the mapper used [
35]. Single-end sequencing is lower in price but faces lower unique mapping rates in repeat-rich regions. For paired-end sequencing, it has to be ensured that the fragment length is more than two times as large as one single read. Otherwise, information is generated twice in the same fragment and the information/price ratio worsens. It has been shown that the error rate could be reduced and the sensitivity enhanced by usage of paired-end bisulfite sequencing [
36]. Therefore, we recommend using paired-end sequencing.
A less cost-intensive approach was set up in the HiSeqXTen [
37], by applying a combination of ten HiSeq systems. Formerly released only for human samples, the system has been opened to other large-scale sequencing experiments of non-human samples (Illumina press release, October 6, 2015). Dependent on the sequencing facility, prices as low as circa 14 €/Gb (2018) were offered.
NovaSeq 6000 is the most recent production scale sequencer and uses the two-color chemistry already shown in the NextSeq system. By combining two nucleotide-binding fluorescence dyes, a four-letter code can be accomplished. A disadvantage of this method is that no signal (zero) codes for one of the four bases within the sequencing process. This lowers the accuracy of the data but offers a faster sequencing process to a much lower price, in the range of HiSeqXTen systems [
38]. Only two wavelength-filtered images of the flow cell need to be computed compared to four images in a four-color chemistry system such as in HiSeq. For WGBS libraries, this technique is not recommended, as a higher GC bias is introduced by bisulfite conversion that could not be tolerated by two-color-chemistry.
Single molecule, real-time sequencing (SMRT-S), such as PacBio and NanoPore sequencing, has shown reliable results for the direct detection of adenine methylation in bacteria based on signal delay of fluorescence-labelled nucleotides [
39] and for 5mC analysis of human DNA [
40]. However, compared to HiSeq or NovaSeq, costs are high (>150 €/Gb). Furthermore, the genome read coverage of such experiments has to be much higher to compensate for the base call error rate of 5–10% while Illumina HiSeq sequencing yields circa 0.0034–1% false base calls [
41]. If these bottlenecks could be solved, the possibility of long reads and, therefore, the facilitated mapping of repeat-rich regions, and the removal of bisulfite treatment from the protocol would be highly advantageous.
4. Multiplexing
To reduce the risk of losing data due to experimental flaws during sequencing, to reach high coverages and read numbers, several samples and types of libraries should be sequenced in combination to yield a high sequencing depth on multiple lanes. A global, equimolar library should be produced based on the quality and quantity (Qubit and/or qPCR measurement) of the DNA sample libraries. The intended sequencing depth or genome coverage is one of the most significant cost factors for WGBS experiments, aside from the number of replicates and the genome size. A sufficient number of reads per covered genome region defines the quality and the statistical power of the downstream analyses. False-positive base calling could be detected more reliably and methylation levels could be determined more accurately at sufficient sequencing depth.
Balancing the coverage and the number of replicates is one of the most important steps within the planning process of WGBS. Ziller et al. [
42] recommended a coverage of 15 fold and three replicates. Beyond that, money would be better spent strengthening the statistical power by increasing the number of statistically independent biological replicates. The theoretical coverage has to be estimated based on the number of homologous chromosomes of the species, the amount of repeats, and the expected degree of heterozygosity, as the level of methylation could differ between the paternal and maternal alleles. For repeat-rich genomes, a coverage of at least 20 fold should be taken into account.
The estimated genome size—not the size of the actual reference assembly—has to be considered for the calculation of total necessary data output as most of the more complex reference genomes assemblies are incomplete, including many scaffolded parts.
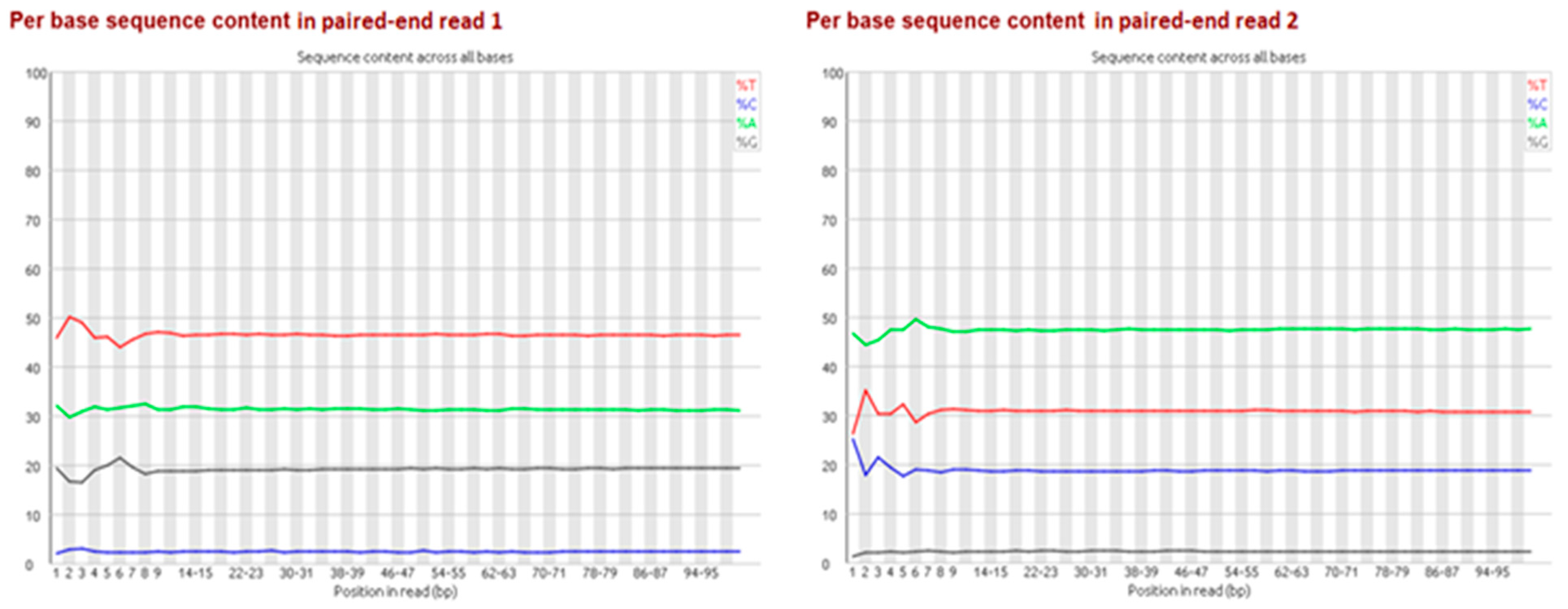
Particularly for bisulfite-treated samples, it is important to multiplex with non-bisulfite-treated libraries, as sequencing technology currently tends to perform worse with extremely unevenly distributed base proportions (e.g., GC-rich or AT-rich libraries). Depending on the rate of methylation, due to bisulfite treatment and the subsequent PCR, generated bisulfite libraries contain a shift in base proportions. The proportion of adenine/thymine is enlarged while the guanine/cytosine proportion is reduced because unmethylated cytosines are converted to thymines via the uracil-intermediates (
Figure 2). For the second paired-end read the proportion of complementary bases shifted because of the bridge amplification during sequencing (
Figure 5). So far, no batch effect between lanes has been reported in literature.
5. Bioinformatics Tools and Benchmarking
An informative overview of the required bioinformatics steps for WGBS after sequencing has been published by Shafi et al. [
43]. Quality check, adapter removal, alignment, methylation calculation, and calculation of differentially methylated regions (DMRs) are the core steps within the bioinformatics pipeline of WGBS experiments (
Figure 3) and could be done by several open-source tools.
As every organism differs in reference genome assembly quality, amount of repeated regions, and homogeneity of base proportion, the applied tools have to be evaluated, ideally by benchmarking them on simulated datasets based on the separate reference genome for every species. The mapper for bisulfite-reads and the DMR caller should be included in such a comprehensive survey.
The alignment tools themselves differ in terms of the algorithms and the strategy used, e.g., three-letter or wild-card alignment, as well as the output file format and content. Some tools report only uniquely mapped reads, whereas others also include multiple-mapped or discarded reads in their output. The alignment tools have to cope with the four different, uncomplementary strands as a result of the combination of bisulfite treatment and PCR, as shown in
Figure 2. The performance of alignment tools differs, depending on the size and amount of repeats in the genome, the coverage, and the sequencing technology used. Hence, for every project the best tools have to be identified and combined to form a data analysis pipeline.
The main difficulty in benchmarking studies is that a known truth has to be generated for multiple class hypothesis testing by the application of simulated datasets. A comprehensive study on simulated and real human lung tumor tissue rrBS data was performed by Sun et al. [
44] using the simulation tool RRBSsim. Here, the tools bwa-meth and BS-Seeker2 showed reliable results for sensitivity, precision, and speed in the mapping of simulated data. For WGBS, no such study was available but, for example, the tool “Sherman—bisulfite-treated Read FastQ Simulator”could be used for the simulation of WGBS datasets based on a given reference genome with parameters like number and size of reads, paired-end or single-end sequencing, conversion rate, number of SNPs, and error rate. Other often used mapping tools are Bismark [
45], BSmap [
46], and Segemehl [
47].
For DMR calling, the available approaches differ in terms of their capability to consider replicates and coverage dependencies, the type of boundary estimation and the statistical tests, suchas logistic regression, binary segmentation, and beta-binomial-based approaches [
41].
The definition of a known truth and the definition of DMRs (e.g., size, coverage, and grade of methylation difference) are also of importance. A small subset of differentially methylated cytosine (DMC) detection tools have been included into a benchmarking analysis by Wreczycka et al. [
48] showing moderate results for methylKit. Better results have been published for metilene [
49] or Defiant [
50]. In subsequent studies, WGBSSuite [
51] could, for example, serve for a simulation of DMRs.
Furthermore, CPU-time, real-time, resident-set-size (rss) and virtual-set-size (vss) memory consumption should be variables to estimate the performance efficiency of the programs.
User friendliness, understood as the degree of installation and handling ease, has to be taken into account for project planning and the benchmarking of bioinformatics tools for the detection of differential methylation by WGBS.
6. Conclusions
We highlighted the current developments in the field of whole-genome bisulfite sequencing, the actual gold standard for 5mC analysis. The application of PCR-free libraries as well as direct methylation detection without bisulfite treatment through SMRT sequencing technologies is seen as a great advantage due to a lower PCR bias or the riddance of bisulfite treatment. However, due to high coverage recommendations and as a result of low base call accuracy, SMRT sequencing for large numbers of samples remains expensive at the moment. In future, this might be solved when lower error rates for these techniques are achieved.
Author Contributions
Conceptualization, C.G. and M.K.; methodology, C.G., M.K., C.B.; investigation, C.G., M.K., C.B.; writing—original draft preparation, C.G. and M.K.; writing—review and editing, M.K., C.B., B.G.; visualization, C.G.; supervision, B.G., I.G.; project administration, C.G., B.G., I.G.; funding acquisition, C.G., B.G., I.G.
Funding
This research was funded by the state of Saxony-Anhalt and Volkswagen Stiftung.
Acknowledgments
We thank Samar Fatma, Alexander Gabel, Martin Posch and Marc Wagner for the valuable scientific discussions as well as Volkswagen Stiftung and the state of Saxony-Anhalt for their financial support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, G.R.; Cohen, S.S. The bases of the nucleic acids of some bacterial and animal viruses. The occurrence of 5-hydroxymethylcytosine. Biochem. J. 1953, 55, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Surani, M.A. DNA methylation dynamics during the mammalian life cycle. Philos. Trans. R. Soc. B 2013, 368, 20110328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Han, Y.; Zhou, L.; Chen, E.; Lu, B.; Liu, Y.; Pan, X.; Cowley, A.W.; Liang, M.; Wu, Q.; et al. A comprehensive evaluation of alignment software for reduced representation bisulfite sequencing data. Bioinformatics 2018, 34, 2715–2723. [Google Scholar] [CrossRef]
- Bock, C.; Tomazou, E.M.; Brinkman, A.B.; Müller, F.; Simmer, F.; Gu, H.; Jäger, N.; Gnirke, A.; Stunnenberg, H.G.; Meissner, A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol. 2010, 28, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.A.; Chan, R.F.; Shabalin, A.A.; Zhao, M.; Turecki, G.; Staunstrup, N.H.; Starnawska, A.; Mors, O.; Xie, L.Y.; van den Oord, E.J. A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics 2017, 12, 743–750. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.-Q.; Ali, I.; Tang, J.; Yang, W.-C. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Stöger, R.; Scaife, P.J.; Shephard, F.; Chakrabarti, L. Elevated 5hmC levels characterize DNA of the cerebellum in Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.M.; Souza, A.L.; Clish, C.B.; Gehring, M. 5-Hydroxymethylcytosine is not present in appreciable quantities in Arabidopsis DNA. G3 (Bethesda, Md.) 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, S.; Wu, Y.; Li, Y.; Chen, T.; Huang, Z.; Liu, S.; Dunwell, T.L.; Pfeifer, G.P.; Dunwell, J.M.; et al. Genome-wide mapping of 5-hydroxymethylcytosine in three rice cultivars reveals its preferential localization in transcriptionally silent transposable element genes. J. Exp. Bot. 2015, 66, 6651–6663. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, F.; Schmidt, M.; van Lijsebettens, M.; Schmidt, T. DNA methylation of retrotransposons, DNA transposons and genes in sugar beet (Beta vulgaris L.). Plant J. 2017, 90, 1156–1175. [Google Scholar] [CrossRef]
- Selker, E.U.; Stevens, J.N. DNA methylation at asymmetric sites is associated with numerous transition mutations. Proc. Natl. Acad. Sci. USA 1985, 82, 8114–8118. [Google Scholar] [CrossRef]
- Lyko, F.; Ramsahoye, B.H.; Jaenisch, R. DNA methylation in Drosophila melanogaster. Nature 2000, 408, 538–540. [Google Scholar] [CrossRef]
- Ausin, I.; Feng, S.; Yu, C.; Liu, W.; Kuo, H.Y.; Jacobsen, E.L.; Zhai, J.; Gallego-Bartolome, J.; Wang, L.; Egertsdotter, U.; Street, N.R.; et al. DNA methylome of the 20-gigabase Norway spruce genome. Proc. Natl. Acad. Sci. USA 2016, 113, E8106–E8113. [Google Scholar] [CrossRef]
- Zhou, J.; Sears, R.L.; Xing, X.; Zhang, B.; Li, D.; Rockweiler, N.B.; Jang, H.S.; Choudhary, M.N.K.; Lee, H.J.; Lowdon, R.F.; et al. Tissue-specific DNA methylation is conserved across human, mouse, and rat, and driven by primary sequence conservation. BMC Genom. 2017, 18, 724. [Google Scholar] [CrossRef] [PubMed]
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-Induced Tissue-Specific Cytosine Methylation Downregulates Expression of HKT Genes in Contrasting Wheat (Triticum aestivum L.) Genotypes. DNA Cell Biol. 2017, 36, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Schmitz, R.J. Covering your bases: Inheritance of DNA methylation in plant genomes. Mol. Plant 2014, 7, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Enomoto, Y.; Dairiki, R.; Ito, T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012, 40, e136. [Google Scholar] [CrossRef] [PubMed]
- Peat, J.R.; Smallwood, S.A. Low Input Whole-Genome Bisulfite Sequencing Using a Post-Bisulfite Adapter Tagging Approach. In DNA Methylation Protocols; Humana Press: New York, NY, USA, 2018; pp. 161–169. [Google Scholar]
- Kint, S.; de Spiegelaere, W.; de Kesel, J.; Vandekerckhove, L.; van Criekinge, W. Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR. PLoS ONE 2018, 13, e0199091. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Okamoto, A. Degradation of DNA by bisulfite treatment. Bioorgan. Med. Chem. Lett. 2007, 17, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Sasaki, T.; Sun, X.; Ma, P.; Lewis, Z.A.; Schmitz, R.J. Methylated DNA is over-represented in whole-genome bisulfite sequencing data. Front. Genet. 2014, 5, 341. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, P.M.; Stirzaker, C.; Song, J.; Grunau, C.; Melki, J.R.; Clark, S.J. Identification and resolution of artifacts in bisulfite sequencing. Methods 2002, 27, 101–107. [Google Scholar] [CrossRef]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- McInroy, G.R.; Beraldi, D.; Raiber, E.-A.; Modrzynska, K.; van Delft, P.; Billker, O.; Balasubramanian, S. Enhanced Methylation Analysis by Recovery of Unsequenceable Fragments. PLoS ONE 2016, 11, e0152322. [Google Scholar] [CrossRef] [PubMed]
- Global Next Generation Sequencing Market Assessment & Forecast. Available online: https://www.prnewswire.com/news-releases/global-next-generation-sequencing-market-assessment--forecast-2017---2021-300431518.html (accessed on 26 November 2018).
- Tran, H.; Porter, J.; Sun, M.-A.; Xie, H.; Zhang, L. Objective and comprehensive evaluation of bisulfite short read mapping tools. Adv. Bioinform. 2014, 2014, 472045. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.; Weng, Z. Evaluation of preprocessing, mapping and postprocessing algorithms for analyzing whole genome bisulfite sequencing data. Brief. Bioinform. 2016, 17, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Luu, P.-L.; Qu, W.; Maddugoda, M.; Huschtscha, L.; Reddel, R.; Chenevix-Trench, G.; Toso, M.; Kench, J.G.; Horvath, L.G.; et al. Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenet. Chromatin 2018. [Google Scholar] [CrossRef] [PubMed]
- Raine, A.; Liljedahl, U.; Nordlund, J. Data quality of whole genome bisulfite sequencing on Illumina platforms. PLoS ONE 2018, 13, e0195972. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Escalona, M.; Rocha, S.; Posada, D. A comparison of tools for the simulation of genomic next-generation sequencing data. Nat. Rev. Genet. 2016, 17, 459–469. [Google Scholar] [CrossRef]
- Ziller, M.J.; Hansen, K.D.; Meissner, A.; Aryee, M.J. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat. Methods 2015, 12, 230–232. [Google Scholar] [CrossRef]
- Shafi, A.; Mitrea, C.; Nguyen, T.; Draghici, S. A survey of the approaches for identifying differential methylation using bisulfite sequencing data. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J.-P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Fast and sensitive mapping of bisulfite-treated sequencing data. Bioinformatics 2012, 28, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Wreczycka, K.; Gosdschan, A.; Yusuf, D.; Grüning, B.; Assenov, Y.; Akalin, A. Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 2017, 261, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. Metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef]
- Condon, D.E.; Tran, P.V.; Lien, Y.-C.; Schug, J.; Georgieff, M.K.; Simmons, R.A.; Won, K.-J. Defiant: (DMRs: easy, fast, identification and ANnoTation) identifies differentially Methylated regions from iron-deficient rat hippocampus. BMC Bioinform. 2018, 19, 31. [Google Scholar] [CrossRef]
- Rackham, O.J.L.; Dellaportas, P.; Petretto, E.; Bottolo, L. WGBSSuite: Simulating whole-genome bisulphite sequencing data and benchmarking differential DNA methylation analysis tools. Bioinformatics 2015, 31, 2371–2373. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




