Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis


Department of Surgery and the Andrew L. Warshaw, MD, Institute for Pancreatic Cancer Research, Massachusetts General Hospital and Harvard Medical School, Boston, 02114 MA, USA
*
Author to whom correspondence should be addressed.
Epigenomes 2018, 2(2), 11; https://doi.org/10.3390/epigenomes2020011
Submission received: 31 May 2018
/
Revised: 14 June 2018
/
Accepted: 15 June 2018
/
Published: 20 June 2018
(This article belongs to the Special Issue Epigenetics of Pancreatic Cancer)
Abstract
:Pancreatic ductal adenocarcinoma has a heterogeneous genetic landscape, marked by frequent mutation of KRAS, CDKN2A, TP53, and SMAD4, resulting in poor responses to conventional therapeutic regimens. Over the past decade, increased understanding of the genetic underpinnings of this lethal cancer has yielded several different characterizations of pancreatic cancer subtypes. However, not all phenotypes and changes in pancreatic cancer can be explained by these findings. New insights on epigenetic modifications associated with pancreatic carcinogenesis have highlighted additional pathways, other than gene mutations, among which chromatin regulation plays a dominant role. Gene expression is highly regulated by subtle changes in chromatin configuration. The underlying mechanism is dominated by reversible post-translational histone modifications. In addition, there is growing evidence that different chromatin mechanisms interact with one another, contributing to the diversity of pancreatic carcinogenesis. This review highlights recent work characterizing chromatin regulatory mechanisms associated with pancreatic carcinogenesis as well as future directions of this emerging research.
Keywords:
pancreatic cancer; epigenetics; chromatin remodeling; histone modification; HAT; HDAC; KDM; BET; SWI/SNF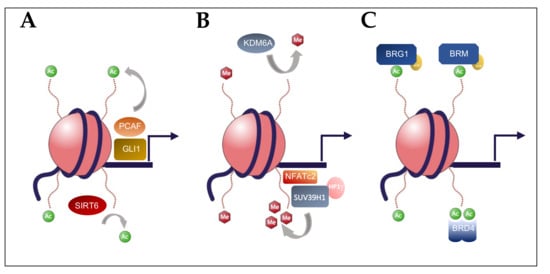
{kind=link}
{kind=link}
{kind=link}
1. Tumor Biology and Epigenetics in PDAC
Pancreatic cancer is an aggressive malignancy associated with poor outcomes and an increasing risk profile [1]. According to current projections, by 2030 it will overtake breast and prostate cancer as the 2nd leading cause of cancer-related death in the Western Hemisphere [2]. Pancreatic ductal adenocarcinoma (PDAC), the most frequent form, is associated with particularly poor prognosis as a result of several factors. First, it is commonly detected in late-stage disease owing to a lack of specific symptoms coupled with aggressive local growth and early metastasis, and second, the clinical picture is accompanied by an extensive desmoplastic stromal reaction that promotes significant resistance to conventional therapeutic options [3]. There are two main precursor lesions which result in PDAC formation (Figure 1). Pancreatic intraepithelial neoplasia (PanIN), which derive from acinar cells undergoing acinar-to-ductal metaplasia (ADM), and larger intraductal papillary mucinous neoplasm (IPMN) of the pancreas [4,5]. Tumors resulting from these precursor lesions have distinct clinicopathological features that impacts tumor biology and prognosis [6]. More than 90% of all PDAC cases harbor KRAS mutations, which are already detectable in these precursor lesions [7]. Moreover, tumor suppressor genes, such as SMAD4, TP53 (commonly referred to as p53), and CDKN2A, are inactivated in 50–70% of all patients [7]. Four different subtypes of pancreatic cancer were recently identified by whole-genome sequencing and copy number variation (CNV) analysis, reflecting the number and location of the gene alterations [8]. Differences in gene expression patterns have similarly been employed to define the transcriptional subtypes of PDAC (Figure 1) [9,10,11]. However, no correlation has been found between these genetic and transcriptional subtypes, suggesting epigenetic mechanisms contribute to these classifications.
Among the post-translational changes in the histone subunit, acetylation and methylation are crucially linked to the carcinogenesis of PDAC. These modifications are regulated by enzymes that add (writers) or remove (erasers) epigenetic marks. Modifications in the acetylation status of histones are accomplished by histone acetyltransferases (HATs) and deacetylases (HDACs) [12]. Common HATs include the CREB-binding protein (CBP), the transcriptional cofactor p300, as well as the p300/CBP-associated factor PCAF, and their activity is associated with active gene expression [13]. In contrast, HDACs generate a more condensed chromatin structure which is associated with gene suppression [14,15]. The balance of these two counterparts creates a finely adjusted balance of (de-) acetylation activity which can be lost during cancer development resulting in tumor enhancement and de-differentiation. Similarly, the methylation status of histone residues results in upregulation or suppression of gene expression through methyltransferases and demethylases [16,17]. In addition, specific “readers”, such as the bromodomain and extraterminal (BET) family of proteins, recognize acetylated histones and recruit additional co-regulators to mediate transcriptional activity in PDAC [18]. Finally, chromatin remodelers, such as SWI/SNF, have ATPase activity that changes the position of the nucleosomes in order to recruit transcriptional machineries to the nucleosomal DNA to regulate gene expression [19]. This review summarizes recent discoveries about how these four broad mechanisms of chromatin regulation play an emerging role in our understanding of PDAC, especially in the development of carcinogenesis from PanIN or IPMN precursor lesions.
2. Histone Acetylation in Pancreatic Carcinogenesis
One of the first studies of the acetylation machinery in PDAC was the description of p300 and its interaction with the nuclear factor of activated T-cells (NFAT) [20]. This study demonstrated that maximal transcriptional activity of Myc in tumors occurs through NFAT-dependent histone acetylation by p300, which allows for the recruitment of an additional sequence-specific DNA binding transcription factor, ELK-1 [20]. More recent work characterized the p300/CREB complex in TGF-β signaling and GLI1-activity in PDAC [21]. In the latter report, it was demonstrated that stimulation of TGF-β signaling resulted in a GLI1-dependent upregulation of a subset of TGF-β responsive genes, including BCL2, IL7, and CCND1. The authors demonstrated a physical and functional association of GLI1 with SMAD2/4 at the BCL2 promoter which regulates BCL2 expression. This GLI/SMAD-dependent activation of TGF-β responsive genes required PCAF (Figure 2A). TGF-β stimulation resulted in a GLI-dependent association of PCAF at the BCL2 promoter with increased acetylation of H3K14, a mark regulated by PCAF. Moreover, depletion of PCAF abrogated TGF-β induced expression of BCL2, IL7, and CCND1. Similar results were recently described in malignant brain tumors, where co-activation of PCAF supports GLI-Hedgehog activity through the acetylation of H3K9 [22].
As a counterpart of the HATs, the deacetylation of histones through HDACs plays a major role in PDAC, and the elevated expression of these enzymes is generally correlated with poor prognosis [23,24]. Early insights on the activity HDACs were found in the regulation of the epithelial-mesenchymal transition (EMT), typified by inactivation of E-cadherin during pancreatic tumorigenesis [25,26,27]. In this work, it was shown that HDACs form complexes with zinc finger proteins ZEB1 and Snail on the promoter of CDH1, the gene encoding E-cadherin, resulting in inactivation of E-cadherin, tumor progression, and metastasis [28,29].
Mutation or loss of the tumor suppressor p53 in PDAC is a common event during pancreatic carcinogenesis and affects almost 50% of all cases [7]. However, not all p53 mutants are inactivated; a subset of these genetic alterations results in gain-of-function mutations that contribute to tumor promotion by enhancing NF-κB activity [30]. A recent study focused on the contribution of HDACs to the regulation of p53 subtypes in PDAC cell lines harboring unique mutations in p53 [15]. It was shown that the expression of p53 mutants in murine and human cells was sensitive to Class I HDAC inhibition. Genetic depletion and chromatin immunoprecipitation experiments revealed that HDAC1 and HDAC2 mediated p53 expression. The authors observed binding of HDAC1 at the promoter of p53, next to the transcriptional start site, as well as in the body of the gene in comparison with HDAC2, which pre-dominantly binds to the p53 promoter. Interestingly, these findings were found in other cancer types, underscoring the regulation of p53 through HDACs [31].
The NAD+-dependent histone deacetylase Sirtuin 6 (SIRT6) is a Class III HDAC with de-acetylation activity towards H3K9 and H3K56 (Figure 2A) [32]. Kugel and colleagues demonstrated in human PDAC specimens that low expression of SIRT6, which represents 30–40% of all cases, correlated with worse survival in 120 PDAC specimens [33]. Employing a GEM mouse model, in which loss of Sirt6 was combined with loss of p53 and activation of Kras (Sirt6f/f;p53f/+;KrasG12D), it was shown that animals lacking Sirt6 had shorter survival and higher metastatic potential. Sirt6 negative mice exhibited hyperacetylation at H3K56, as well as an increase of chromatin-bound Myc compared to wild-type littermates. Taking a closer look at the Sirt6 KO phenotype, ChIP-seq experiments identified Lin28b as one of the top genes with increased H3K56Ac at its transcription start side which correlated with an upregulation of Lin28b in SIRT6-deficient PDAC cells. Interestingly, expression of Lin28b is usually restricted to embryonic tissues [34] undergoing re-activation through tumorigenesis in different tissues [35,36,37]. Through chromatin immunoprecipitation the authors demonstrated higher levels of H3K56Ac and H3K9Ac at MYC binding sites at the Lin28b promoter in SIRT6-negative cells, reflecting an open chromatin confirmation. Furthermore, knockdown of SIRT6 in SIRT6high PDAC cells resulted in prompt acetylation of these epigenetic marks. Suppression of Myc also led to a reduction in the expression of Lin28b, supporting the idea of antagonism between SIRT6 and Myc. Further functions of Lin28b were explored and demonstrated a role for Lin28b in the inhibition of the tumor suppressor micro-RNA family members let-7 [38]. let-7 members by themselves inhibit target genes (IGF2BPs, HMGA2) which drive carcinogenesis [39,40] and here were demonstrated to be overexpressed in SIRT6low PDAC cells. Taken together, these results demonstrate the hyperacetylation of H3K9Ac and H3K56Ac through loss of SIRT6, which opens Myc binding sides at the Lin28b promoter region. Ultimately, Lin28b inhibits the tumor suppressor let-7 micro-RNA family by upregulating the metabolic target genes (IGF2BPs, HMGA2) represented in a subpopulation of PDAC patients with worse overall survival.
3. Histone Methylation in Pancreatic Carcinogenesis
The methylation status of lysine residues on histone tails can have both positive and negative effects on gene expression. While some histone methylation marks are strongly associated with active (H3K4me3) or repressed (H3K27me3) transcription, the function of many of these modifications is context dependent [41]. A role for histone methylation and formation of heterochromatin in PDAC was found in the silencing of tumor suppressor CDKN2B (p15, Ink4b), a frequent event in PDAC [42]. This study defined a key role for NFATc2 in establishing transcriptional silencing of p15 (Figure 2B). Co-immunoprecipitation and ChIP experiments revealed that NFATc2 physically interacts with the histone methyltransferase SUV39H1 which allows for its NFATc2-dependent recruitment to the p15 promoter. The NFATc2-SUV39H1 complex results in trimethylation of H3K9, a suppressive epigenetic mark, and decreased p15 expression. The suppressive effects of the NFATc2-SUV39H1 complex were mediated by the recruitment of HP1γ, a heterochromatin protein family member which stabilizes transcription complexes that contribute to heterochromatin expansion [43,44,45]. Depletion of either NFATc2 or SUV39H1 resulted in loss of HP1γ binding to the p15 promoter. Moreover, NFATc2-SUV39H1 complex formation was stabilized in the presence of HP1; depletion of HP1 resulted in a de-stabilization of the repressor complex with a loss of p15 promoter silencing through NFATc2 [42].
Enhancer of Zeste Homologue (EZH2) has been implicated in inactivating the expression of tumor suppressor genes in a variety of cancers by mediating the tri-methylation of H3K27 [46,47]. An early study in PDAC defined an important role for EZH2 in silencing the tumor suppressor CDKN1B (p27) in poorly differentiated PDACs [48]. More recently, Chen and colleagues described the context-specific roles of EZH2 in the regulation of NFATc1 expression during pancreatic regeneration and tumorigenesis [49]. Employing pancreatitis models in mice, the authors found that NFATc1 was activated in acinar cells after injury and was silenced by EZH2-mediated H2K27me3 at its promoter during the late stages of regeneration. Dysregulation of this mechanism by genetic loss of EZH2 or ectopic expression of NFATc1 prevented proper regeneration of the pancreas. Interestingly, the effects of EZH2 on NFATc1 expression were quite different against the background of pancreatic expression of KrasG12D. Genetic depletion or pharmacologic inhibition of EZH2 resulted in a decrease in NFATc1 expression in murine-derived PDAC cells (KrasG12D;p53R172/+). ChIP experiments revealed that loss of EZH2 activity corresponded with a decrease in H3K4me3 at the transcription start site of the NFATc1 promoter. These positive regulatory effects of EZH2 on NFATc1 were not observed in the PRC2 target gene Hoxa10, suggesting that oncogenic activation of Kras selectively alters the activity of EZH2 on a subset of target genes. These results highlight the plasticity of histone-modifying enzymes in the regulation of regeneration and tumorigenesis [49].
Lysine-specific histone demethylases (KDMs) have both tumor promoting and suppressive functions in human cancer [50,51]. The lysine-specific histone demethylase 1A (LSD1), also known as KDM1A, is overexpressed in PDAC and silencing of LSD1 resulted in an impaired glucose uptake, decreased cell viability, and reduced tumor growth in vivo [52]. Underscoring this putative mechanism, it was shown that LSD1 stabilizes HIF-1α expression which maintains glucose metabolism in hypoxic tumor environments [52]. An additional study investigating the role of the H3K36 demethylase KDM2B in PDAC revealed that silencing of KDM2B resulted in impaired growth of PDAC cell lines [53]. Further in vivo studies highlighted the role of KDM2B in tumor growth and poor differentiation in mice with pancreatic expression of KrasG12D. In addition, the authors emphasized the contribution of KDM2B in Myc activity together with KDM5A and EZH2. These findings link KDM2B to an aggressive PDAC subtype with integral roles in Myc transcriptional activity [53].
A recent study has identified a role for KDM6A (UTX), an X chromosome-encoded H3K27me3 demethylase, in PDAC subtype specification [54]. Independent of its demethylase activity, KDM6A is part of the COMPASS (complex of proteins associated with SET1)-like complex which mediates monomethylation of H3K4 to establish active enhancers [55,56]. The authors described frequent mutations and deletions of KDM6A in PDAC tumors with a squamous-like subtype transcriptional signature (Figure 2B) [11]. KDM6A exhibited strong expression in PanIN lesions and well-differentiated PDAC, whereas its expression was absent in poorly differentiated PDAC and metastatic lesions. In line with these expression patterns in human PDAC, tumors from female Kdm6a null mice with a KrasG12D background exhibited histologic features and gene expression patterns reflecting squamous differentiation. Interestingly, only a minor subset of differentially expressed genes in Kdm6a null mice carried elevated levels of H3K27me3 marks, indicative of demethylase-independent changes of KDM6A deficiency. Consistent with its role as a component of COMPASS-like complex, the authors demonstrated loss of KDM6A disrupts COMPASS-like complex formation at a subset of super enhancers, resulting in the aberrant activation of genes involved in squamous differentiation including Tp63, Myc, and Runx3. Both human and murine pancreatic cancer cell lines deficient in KDM6A were more sensitive to inhibitors of the BET proteins, a family of epigenetic readers (described below) [57]. Strikingly, the BET family member BRD4 is bound to super enhancers activated by loss of KDM6A (ΔNp63 and Runx3) and BET inhibition through JQ1 downregulates their expression. Together these recent findings emphasize the importance of super enhancers in PDAC progression and the need of re-programming these subtypes through additional therapies [54].
4. BET Bromodomain Regulation in Pancreatic Carcinogenesis
The bromodomain and extra-terminal (BET) family have emerged as key epigenetic contributors to PDAC. BET proteins (BRD2, BRD3, BRD4, and the testis-restricted BRD-T) contain tandem bromodomains that mediate binding to acetylated lysines on histone and non-histone substrates [58]. BET family members function as chromatin adaptors that recruit the super elongation complex (SEC) and polymerase-associated factor complex (PAFc) to target genes to mediate transcriptional elongation of RNA [18]. In humans, the expression of BRD2 and BRD3 is elevated during the early histological stages of tumorigenesis (ADM and PanIN lesions), with high levels of BRD2, 3, and 4 found in tumors [57]. BET proteins are similarly induced in a genetically engineered mouse (GEM) model with pancreas-specific expression of KrasG12D, and pharmacological inhibition of BET bromodomains blocks ADM and PanIN formation in mice [59]. Contemporaneous studies employing RNAi targeting of specific BET family members and small molecules that inhibit the binding of BET bromodomains have defined a critical role for BET proteins in the growth of PDAC cells in vitro, as well as the in vivo growth of patient-derived xenograft (PDX) and GEM models of PDAC [57,59,60,61,62].
BET proteins contribute to PDAC by mediating the activity of several key transcriptional programs. Much of the early excitement concerning BET proteins was based on their ability to directly regulate the expression of MYC. MYC amplification is detected in about 14% of PDAC and is enriched in the squamous subtype [11,63]. Consistent with this finding, gene expression analysis of more than 55 PDX models of PDAC identified 30% that had high expression of MYC target genes [64]. Furthermore, tumors from these MYChigh patients were more proliferative, less differentiated, and associated with shorter overall survival. Importantly, cell culture and tumor models revealed that MYChigh PDACs were more sensitive to BET bromodomain inhibition than those derived from MYClow tumors. However, it remains unclear to what extent the regulation of MYC by BET proteins contributes to these effects. Several studies have found MYC expression to be regulated by BET proteins in a subset of PDAC cell lines; however, the BET-dependent expression of MYC and BET-dependent growth of PDAC cells frequently do not correlate [57,59]. Moreover, depletion of MYC only partially recapitulates the effects of BET bromodomain inhibition, supporting a role for other BET-dependent pathways in PDAC. Among these are additional transcriptional programs including STAT3 and GLI [59]. BET proteins regulate the activity of STAT3 in PDAC cells through transcriptional regulation of its upstream activator IL6. Pharmacological inhibition of BET bromodomains resulted in loss of BRD4 binding to IL6 promoter, decreased expression of IL6, and reduced levels of active pSTAT3 in both human PDAC cell lines and a GEM model (Ptf1a-Cre; KrasG12D; p53fl/fl) of PDAC (Figure 2C). Importantly, exogenous administration of IL6 in GEM mice restored pSTAT3 levels and abrogated the inhibitory effects of BET bromodomain inhibition on tumor growth [59]. One of the dominant gene programs altered after BET bromodomain inhibition in PDAC cells is regulated by GLI [57]. In contrast to other transcriptional programs regulated by BET proteins in PDAC, GLI represents a growing body of sequence-specific DNA-binding proteins that physically interact with BET proteins. Co-immunoprecipitation experiments demonstrated that BRD2, BRD3, and BRD4 interact with GLI1 and GLI2. Pharmacological inhibition of BET bromodomains disrupts BRD4-GLI1 complexes, resulting in diminished GLI transcriptional activity in PDAC cells [57]. A link between BET and GLI in PDAC was further revealed by a study demonstrating that PDAC cells with acquired resistance to BET bromodomain inhibition are dependent on the elevated expression of GLI2 for this resistance [65].
A hallmark of pancreatic cancer is a highly desmoplastic stroma, composed of abundant fibrosis and cancer-associated fibroblasts (CAFs) [66,67]. There has been increased appreciation for the impact of cross-talk between neoplastic and stromal cells on PDAC tumor biology. Soluble factors secreted by these cell populations play a large role in mediating this cross-talk. A recent study demonstrated that PDAC cells exhibited altered metabolic activity and expression of genes involved in metabolic pathways when exposed to conditioned media from immortalized human CAF cultures [68]. These changes corresponded to a rapid increase in global acetylation of H3K9 and H3K27, and these epigenetic marks were enriched in the promoters and enhancers of genes that were responsive to CAF-conditioned media. Consistent with this increased acetylation, the response of these genes to conditioned media was found to be dependent on the activity of BET proteins. Interestingly, while BRD2 and BRD4 have been identified as regulators of BET target genes in PDAC cells, BRD2 was uniquely responsible for the gene expression changes induced by CAF-conditioned media [68]. The activity of BET proteins within PDAC cells also contributes to the tumor microenvironment. BET proteins regulate the expression of SHH in human PDAC cells and a GEM model (Pdx-1-Cre;KrasG12D;p53fl/fl) of PDAC. SHH secreted by PDAC cells functions via paracrine signaling to recruit and activate CAFs in tumors. Consistent with this activity, BET bromodomain inhibition and shRNA mediated depletion of BRD2 or BRD4 results in reduced SHH expression and diminished CAF content in PDAC tumors [57].
In addition to their contribution to PDAC-mediated activation of CAFs, BET proteins directly contribute to the biology of CAFs [62]. While CAFs are a diverse population of cells, the majority are believed to derive from resident quiescent pancreatic stellate cells [67]. Within the tumor microenvironment these cells become activated (alpha smooth muscle actin (α-SMA)-positive) and express elevated levels of collagen, which contributes to the fibrotic stroma. Consistent with a role for BET proteins in the activation of CAFs, BET bromodomain inhibition decreased α-SMA levels in primary and immortalized cultures of human CAFs, without affecting the viability of these cells. Moreover, upon inhibition of BET proteins, CAFs exhibited a downregulation of ECM-related genes (COL1A1, COL1A2, COL1A3 and FN1) [62]. Taken together, BET proteins are crucially involved in the carcinogenesis of PDAC. This is not only true for their contribution to early cancer progression through PanIN lesions but also for their various interactions in stromal crosstalk through CAFs which drive fibrotic reactions in PDAC.
5. Chromatin Remodelers (SWI/SNF) in Pancreatic Carcinogenesis
In past years, insights into chromatin remodelers highlighted their diverse context-dependent functions in pancreatic carcinogenesis. Research in this field has been predominantly performed on SWI/SNF enzymes which drive chromatin remodeling through their ATPase subunits BRG1 and BRM [19,69]. SWI/SNF proteins recognize specific histone marks, and through dynamic ATP-dependent modifications, allow the recruitment of transcriptional machineries to the nucleosomal DNA (Figure 2C). Mutations in SWI/SNF subunits, which are found in approximately 20% of human cancers, lead to cell cycle defects and the promotion of tumor formation [70]. In one of the first studies to characterize the SWI/SNF components in PDAC, Numata et al. found that high expression of BRM was correlated with poor survival [71]. Another study found that patients with a germ-line polymorphism in BRM (BRM-741, BRM-1321) exhibited worse survival compared to patients with wild-type BRM [72]. A recent study suggested that regulation of the JAK/STAT pathway is a major contribution of the BRG1/BRM subunits in PDAC [73]. The authors observed cell cycle arrest and suppression of tumor growth after BRM silencing in vivo. In addition, downstream targets of the JAK2/STAT3 pathway (CCND1, Survivin, MMP7, and VEGF) were decreased after silencing BRM. In line with these findings, levels of active pSTAT3 were downregulated and could be restored with exogenously supplied IL6 [73].
Taking a closer look at BRG1, Molin et al. discovered unique insights on the expression patterns in IPMNs [74]. More than 50% of all IPMN specimens exhibited reduced expression of BRG1 when compared to normal tissue. In addition, high-grade IPMNs more frequently had lower BRG1 levels, suggesting that loss of BRG1 drives de-differentiation. A subsequent study demonstrated additional context-dependent functions of BRG1 in the development of different PDAC subtypes or its precursor lesions [75]. First, it was shown that loss of Brg1 in KrasG12D-driven mice resulted in formation of cystic neoplasms which resembled human IPMNs. In a time-dependent manner these precursor lesions were able to form invasive cancer. However, compared to mice with an ordinary KrasG12D-driven PanIN, these lesions were less proliferative and had a decreased gene expression signature for pathways regulating cellular motility, invasion, and metastasis. Further investigation of downstream pathways revealed that tumors from Brg1-negative mice almost completely lost the expression of tumor-suppressive (p53, p21, and p16) and tumor-promoting genes (Hmga2), when compared to ordinary PDACs. In line with these findings, the promoter of Hmga2 showed an enrichment of BRG1 accompanied by the repressive histone mark H3K27me3. Interestingly, the authors demonstrated that loss of BRG1 has cell context specific effects in the formation of pre-neoplastic lesions. GEM models that allowed for pancreatic acinar or ductal cell-specific deletion of Brg1 revealed BRG1 expression was required for KrasG12D-driven PanIN formation from adult acinar cells, whereas loss of Brg1 was required for KrasG12D-driven IPMN formation from adult pancreatic duct cells [75]. In a follow-up study from the same group it was shown that BRG1 promotes tumorigenesis in PDAC through an EMT-like gene expression pattern; whereas it inhibits de-differentiation in adult pancreatic duct cells and blocks neoplastic transformation [76]. Taken together, these findings accentuate the importance of Brg1 as a stage-dependent chromatin remodeler with distinct functions during pancreatic carcinogenesis.
In addition to BRG1 and BRM, ARID1a contributes to SWI/SNF function by mediating promoter occupancy [77]. In addition to being frequently mutated in PDAC [7], Arid1a deletion leads to the formation of colon cancer in mice [78] and drives ovarian carcinogenesis through mTOR activation [79,80]. Kimura and colleagues investigated the role of ARID1a and BRG1 in pancreatic tumorigenesis [81]. Employing KrasG12D-driven mice lacking Arid1a they demonstrated that mice deficient in Arid1a developed IPMNs and PDAC more frequently when compared to ordinary KrasG12D or Arid1afl/+ mice, thereby documenting the tumor suppressive abilities of Arid1a in PDAC. Tumors from Arid1afl/fl mice exhibited an upregulation of pSTAT3 and a similar suppression of p53, p21, and p16 as described above for Brg1-deficient mice [75]. The authors investigated further key signaling pathways and found a correlation with an increase in mTOR activity in Arid1afl/fl mice. Interestingly, these effects were not detectable in Brg1-deficient mice, highlighting a unique role for IPMN-associated PDAC formation through mTOR signaling in the absence of Arid1a [81]. Taken together, histone remodelers reassemble a highly heterogeneous group that controls pancreatic carcinogenesis through their different subgroups. They play an important role in tumor progression from ordinary PanIN formations and IPMN-associated PDAC. Notably, SWI/SNF subunits act in a stage-dependent manner with different contributions on tumor formation in PDAC.
6. Outlook
Alterations in chromatin regulation and their contribution to key pathways in PDAC has become an important field of research. Along with recent studies on the acetylation or methylation status of histones, chromatin readers and remodelers implement context-specific functions in PDAC. In particular, stage-dependent activities of different chromatin regulators elucidate the complex regulatory machinery behind pancreatic carcinogenesis. Furthermore, recent findings emphasize the role of chromatin mechanisms in cancer–stroma crosstalk, opening multiple directions for future studies and therapeutic strategies.
Funding
Thomas Hank is funded by a Mildred-Scheel-Postdoctoral Fellowship from German Cancer Aid.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-del Castillo, C.; Adsay, N.V. Intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2010, 139, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.T.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of SOX9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of precursor lesions for pancreatic cancer: The genetics and biology of intraductal papillary mucinous neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Grimmond, S.M.; Biankin, A.V. Pancreatic cancer genomics. Curr. Opin. Genet. Dev. 2014, 24, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, N.; Goodman, R.H. Creb-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schafer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of bet recruitment to chromatin as an effective treatment for mll-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Linhart, T.; Schlengemann, K.; Reutlinger, K.; Wegele, J.; Adler, G.; Singh, G.; Hofmann, L.; Kunsch, S.; Buch, T.; et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology 2010, 138, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Nye, M.D.; Almada, L.L.; Fernandez-Barrena, M.G.; Marks, D.L.; Elsawa, S.F.; Vrabel, A.; Tolosa, E.J.; Ellenrieder, V.; Fernandez-Zapico, M.E. The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J. Biol. Chem. 2014, 289, 15495–15506. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone acetyltransferase PCAF is required for hedgehog-GLI-dependent transcription and cancer cell proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Kramer, O.H.; Schmid, R.M.; Saur, D. Acetylation as a transcriptional control mechanism-HDACS and HATS in pancreatic ductal adenocarcinoma. J. Gastrointest. Cancer 2011, 42, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed e box binding zinc finger protein SIP1 downregulates e-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Goder, A.; Beyer, M.; Kiweler, N.; Mahendrarajah, N.; Rauch, A.; Nikolova, T.; Stojanovic, N.; Wieczorek, M.; Reich, T.R.; et al. Class I histone deacetylases regulate p53/NF-ĸb crosstalk in cancer cells. Cell. Signal. 2017, 29, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Kugel, S.; Sebastian, C.; Fitamant, J.; Ross, K.N.; Saha, S.K.; Jain, E.; Gladden, A.; Arora, K.S.; Kato, Y.; Rivera, M.N.; et al. SIRT6 suppresses pancreatic cancer through control of lin28b. Cell 2016, 165, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.G.; Tang, L. Conservation of the heterochronic regulator lin-28, its developmental expression and microrna complementary sites. Dev. Biol. 2003, 258, 432–442. [Google Scholar] [CrossRef]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. Lin28b promotes colon cancer progression and metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Katsaros, D.; Shaverdashvili, K.; Qian, B.; Wu, Y.; de la Longrais, I.A.; Preti, M.; Menato, G.; Yu, H. Pluripotent factor lin-28 and its homologue lin-28b in epithelial ovarian cancer and their associations with disease outcomes and expression of let-7a and IGF-II. Eur. J. Cancer 2009, 45, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O'Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 mediates the terminal uridylation of let-7 precursor microrna. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Park, S.M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and HMGA2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. PRB family proteins are required for H3K27 trimethylation and polycomb repression complexes binding to and silencing P16INK4α tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.; Glesel, E.; Singh, G.; Chen, N.M.; Reutlinger, K.; Zhang, J.; Billadeau, D.D.; Fernandez-Zapico, M.E.; Gress, T.M.; Singh, S.K.; et al. Restricted heterochromatin formation links NFATC2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology 2012, 142, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Workman, J.L. The heterochromatin protein 1 (HP1) family: Put away a bias toward HP1. Mol. Cells 2008, 26, 217–227. [Google Scholar] [PubMed]
- Gilbert, N.; Boyle, S.; Sutherland, H.; de Las Heras, J.; Allan, J.; Jenuwein, T.; Bickmore, W.A. Formation of facultative heterochromatin in the absence of HP1. EMBO J. 2003, 22, 5540–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, A.L.; Oulad-Abdelghani, M.; Ortiz, J.A.; Remboutsika, E.; Chambon, P.; Losson, R. Heterochromatin formation in mammalian cells: Interaction between histones and HP1 proteins. Mol. Cell 2001, 7, 729–739. [Google Scholar] [CrossRef]
- Abbosh, P.H.; Montgomery, J.S.; Starkey, J.A.; Novotny, M.; Zuhowski, E.G.; Egorin, M.J.; Moseman, A.P.; Golas, A.; Brannon, K.M.; Balch, C.; et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res. 2006, 66, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ochiai, A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008, 99, 738–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.M.; Neesse, A.; Dyck, M.L.; Steuber, B.; Koenig, A.O.; Lubeseder-Martellato, C.; Winter, T.; Forster, T.; Bohnenberger, H.; Kitz, J.; et al. Context-dependent epigenetic regulation of nuclear factor of activated T cells 1 in pancreatic plasticity. Gastroenterology 2017, 152, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, L.; Emre, N.C.; Kruhlak, M.J.; Chung, H.J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhu, W.; Xu, W.; Zhang, B.; Shi, S.; Ji, S.; Liu, J.; Long, J.; Liu, C.; Liu, L.; et al. LSD1 sustains pancreatic cancer growth via maintaining HIF1α-dependent glycolytic process. Cancer Lett. 2014, 347, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Paskaleva, P.; Ferrari, F.; Deshpande, V.; Stoykova, S.; Contino, G.; Wong, K.K.; Lan, F.; Trojer, P.; Park, P.J.; et al. KDM2B promotes pancreatic cancer via polycomb-dependent and -independent transcriptional programs. J. Clin. Investig. 2013, 123, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6α activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to bet inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Tang, Z.; Chen, C.W.; Shimada, M.; Koche, R.P.; Wang, L.H.; Nakadai, T.; Chramiec, A.; Krivtsov, A.V.; Armstrong, S.A.; et al. A UTX-MLL4-p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol. Cell 2017, 67, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.S.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI underlies a role for bet bromodomains in pancreatic cancer growth and the tumor microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of bet family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. Bet bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The bet bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the bet bromodomain inhibitor jq1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the bet bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the bet bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cote, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Morinaga, S.; Watanabe, T.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Nakamura, Y.; Kameda, Y.; Okawa, S.; Rino, Y.; et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Segedi, M.; Anderson, L.N.; Espin-Garcia, O.; Borgida, A.; Bianco, T.; Cheng, D.; Chen, Z.; Patel, D.; Brown, M.C.; Xu, W.; et al. BRM polymorphisms, pancreatic cancer risk and survival. Int. J. Cancer 2016, 139, 2474–2481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.; Du, C.; Guo, H.; Ma, L.; Liu, X.; Kornmann, M.; Tian, X.; Yang, Y. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017, 402, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Molin, M.D.; Matthaei, H.; Wu, J.; Blackford, A.; Debeljak, M.; Rezaee, N.; Wolfgang, C.L.; Butturini, G.; Salvia, R.; Bassi, C.; et al. Clinicopathological correlates of activating gnas mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann. Surg. Oncol. 2013, 20, 3802–3808. [Google Scholar] [CrossRef] [PubMed]
- Von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris Iv, J.P.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.L.; Brennan, J.; Schisler, J.C.; Serber, D.; Patterson, C.; Magnuson, T. Arid1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell Biol. 2013, 33, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Rahmanto, Y.S.; Wu, R.C.; Wang, Y.; Wang, Z.; Wang, T.L.; Shih, I.M. Roles of deletion of arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.L.; Damrauer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 2015, 6, 6118. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Fukuda, A.; Ogawa, S.; Maruno, T.; Takada, Y.; Tsuda, M.; Hiramatsu, Y.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. ARID1A maintains differentiation of pancreatic ductal cells and inhibits development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2018, in press. [Google Scholar] [CrossRef] [PubMed]
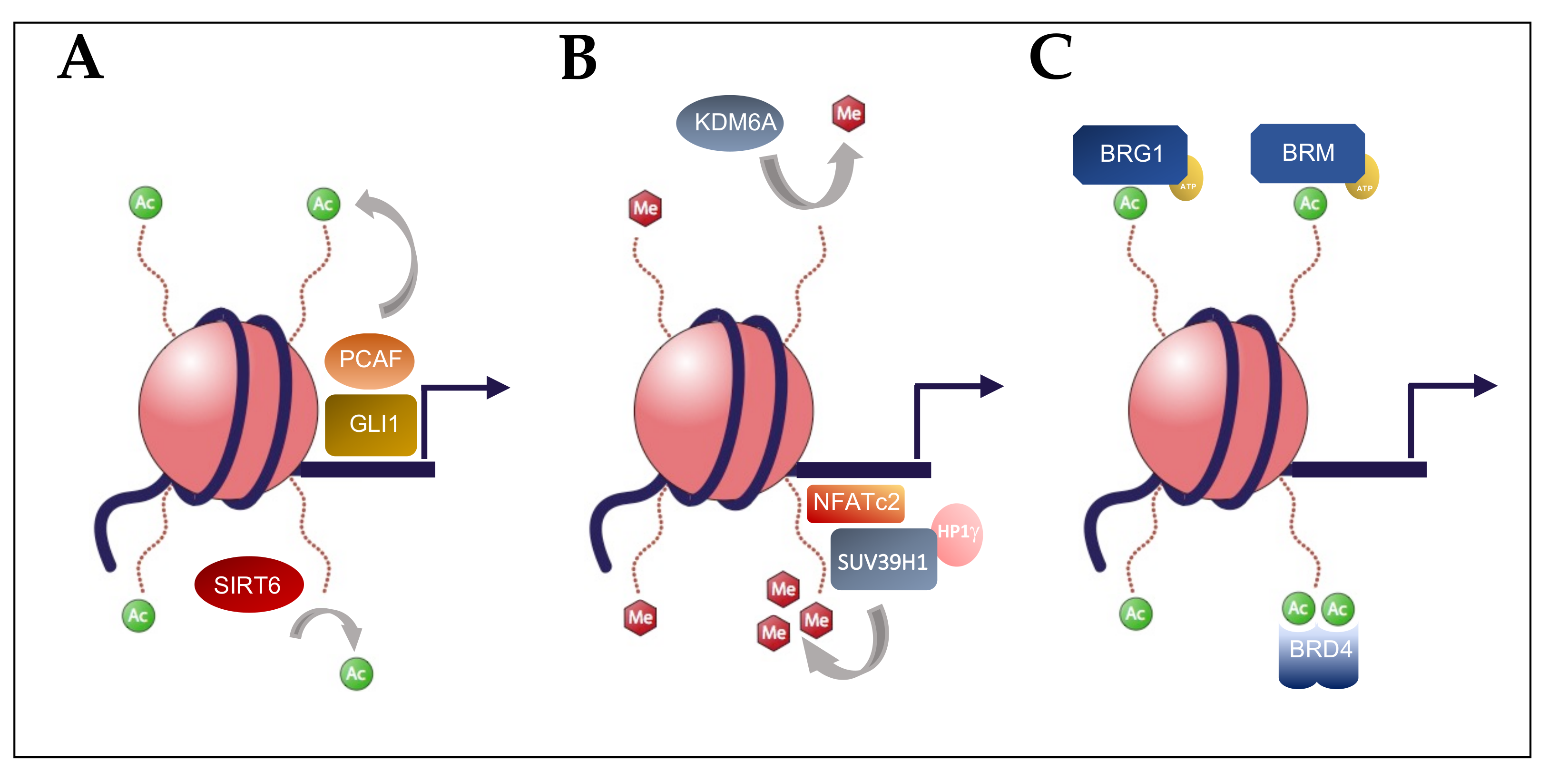
Figure 1.
Precursor lesions of pancreatic cancer. Pancreatic acinar cells can acquire a duct-like phenotype through a process termed acinar-to-ductal metaplasia (ADM). In the context of activating mutations in KRAS, these lesions can progress to pancreatic intraepithelial neoplasia (PanIN; top). Pancreatic ductal cells with activating mutations to KRAS and GNAS form a second class of precursor lesions, intraductal papillary mucinous neoplasms (IPMN, bottom). Subsequent acquisition of inactivating tumor suppressor genes in PanIN and IPMN lesions result in the progression to PDAC. Transcriptional analysis of tumors defined different PDAC subtypes (right) [12].
Figure 1.
Precursor lesions of pancreatic cancer. Pancreatic acinar cells can acquire a duct-like phenotype through a process termed acinar-to-ductal metaplasia (ADM). In the context of activating mutations in KRAS, these lesions can progress to pancreatic intraepithelial neoplasia (PanIN; top). Pancreatic ductal cells with activating mutations to KRAS and GNAS form a second class of precursor lesions, intraductal papillary mucinous neoplasms (IPMN, bottom). Subsequent acquisition of inactivating tumor suppressor genes in PanIN and IPMN lesions result in the progression to PDAC. Transcriptional analysis of tumors defined different PDAC subtypes (right) [12].
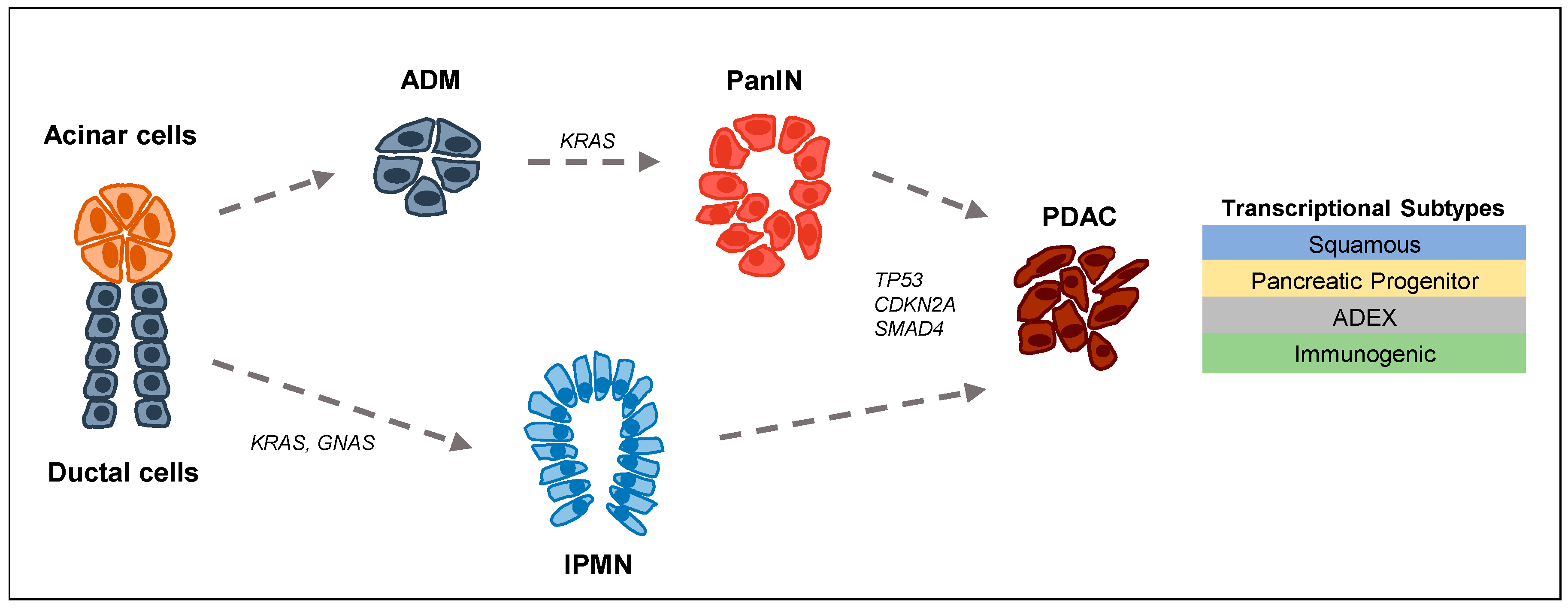
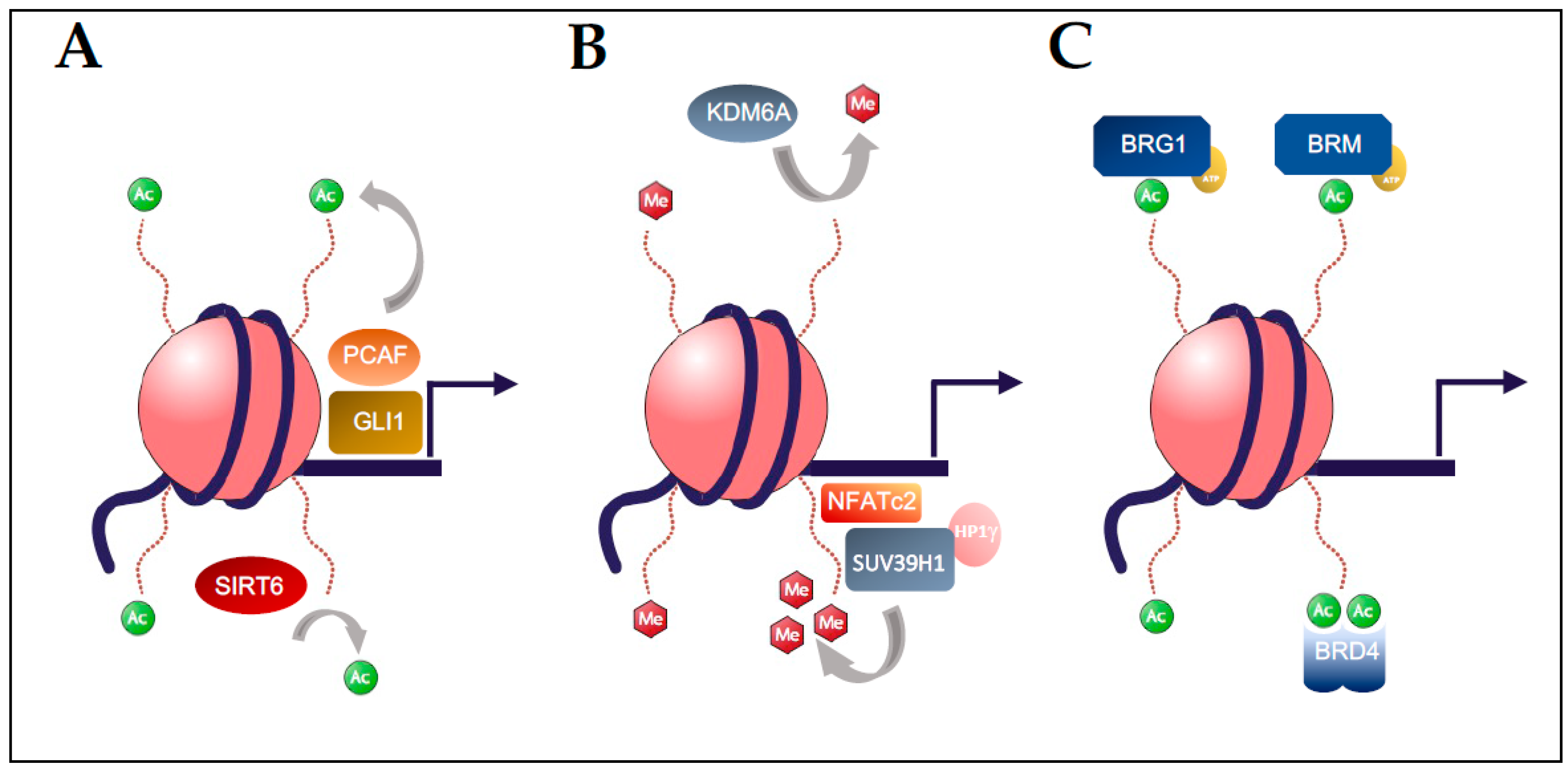
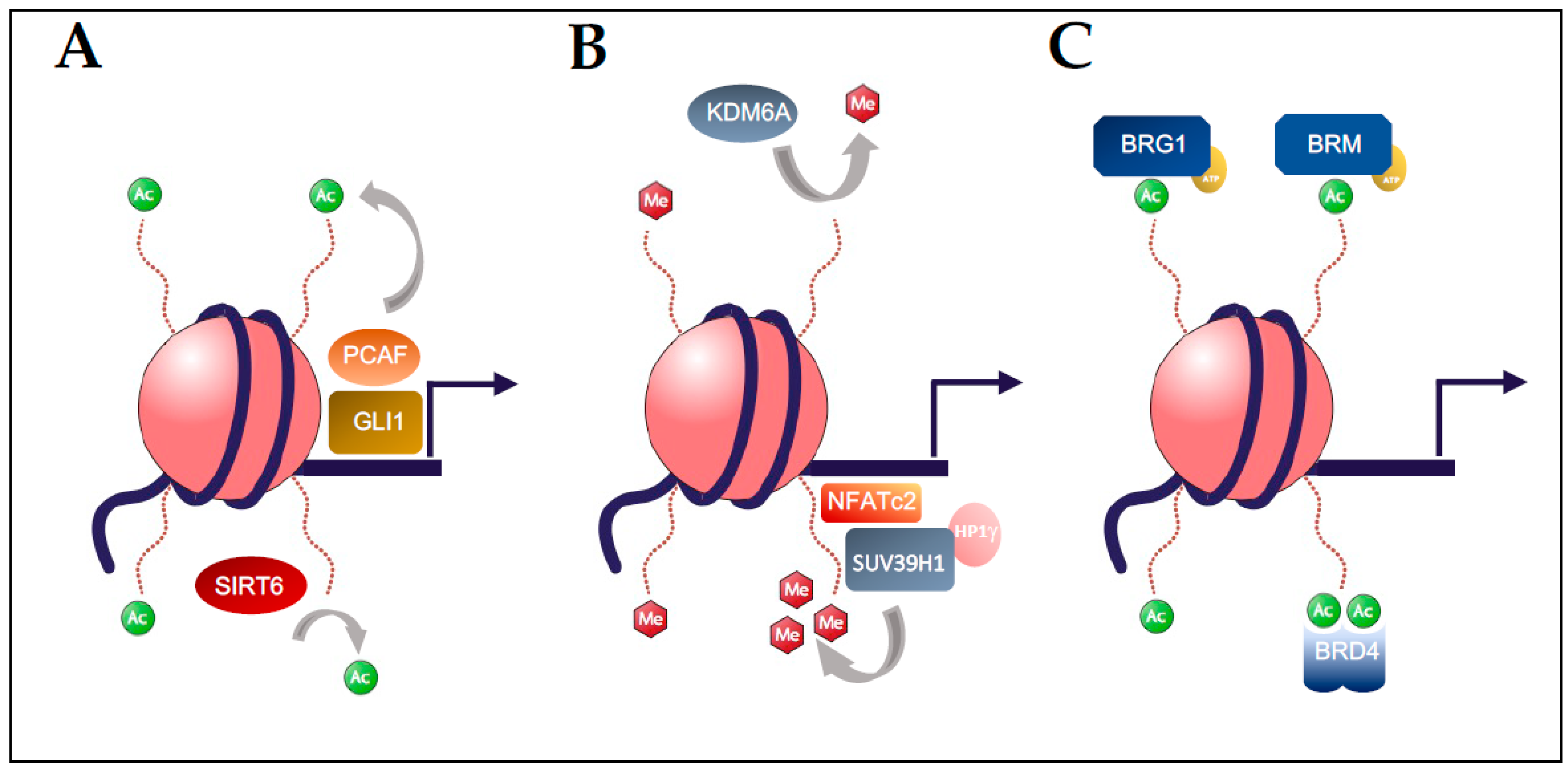
Figure 2.
Post-translational modifications of chromatin regulate gene expression in pancreatic carcinogenesis. (A) Histone acetylation is mediated by the p300/CREB associated complex (PCAF) which is regulated by binding of GLI1 and results in acetylation of H3K14 to increase expression of a subset of TGF-β responsive genes. Similarly, recruitment of the HDAC SIRT6 to promoters results in deacetylation of H3K9 and H3K56 with suppression of pro-tumorigenic gene programs. (B) The histone methyltransferase SUV39H1 can bind to target promoters by forming a complex with DNA-bound NFATc2, leading to gene suppression through trimethylation of H3K9 and recruitment of the heterochromatin protein family member HP1γ. In contrast, loss of the H3K27 demethylase KDM6A promotes PDAC with a squamous differentiation. (C) SWI/SNF chromatin remodelers have distinct functions in PDAC that are mediated by their ATP-dependent subunits BRG1 and BRM, which contribute to IL6/STAT3 signaling in PDAC. Likewise, specific epigenetic readers, such as BRD4, recognize acetylation marks and regulate a variety of transcriptional programs in PDAC, including the IL6/STAT3 axis.
Figure 2.
Post-translational modifications of chromatin regulate gene expression in pancreatic carcinogenesis. (A) Histone acetylation is mediated by the p300/CREB associated complex (PCAF) which is regulated by binding of GLI1 and results in acetylation of H3K14 to increase expression of a subset of TGF-β responsive genes. Similarly, recruitment of the HDAC SIRT6 to promoters results in deacetylation of H3K9 and H3K56 with suppression of pro-tumorigenic gene programs. (B) The histone methyltransferase SUV39H1 can bind to target promoters by forming a complex with DNA-bound NFATc2, leading to gene suppression through trimethylation of H3K9 and recruitment of the heterochromatin protein family member HP1γ. In contrast, loss of the H3K27 demethylase KDM6A promotes PDAC with a squamous differentiation. (C) SWI/SNF chromatin remodelers have distinct functions in PDAC that are mediated by their ATP-dependent subunits BRG1 and BRM, which contribute to IL6/STAT3 signaling in PDAC. Likewise, specific epigenetic readers, such as BRD4, recognize acetylation marks and regulate a variety of transcriptional programs in PDAC, including the IL6/STAT3 axis.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hank, T.; Liss, A.S. Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis. Epigenomes 2018, 2, 11. https://doi.org/10.3390/epigenomes2020011
AMA Style
Hank T, Liss AS. Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis. Epigenomes. 2018; 2(2):11. https://doi.org/10.3390/epigenomes2020011
Chicago/Turabian StyleHank, Thomas, and Andrew S. Liss. 2018. "Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis" Epigenomes 2, no. 2: 11. https://doi.org/10.3390/epigenomes2020011