Panobinostat Potentiates Temozolomide Effects and Reverses Epithelial–Mesenchymal Transition in Glioblastoma Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
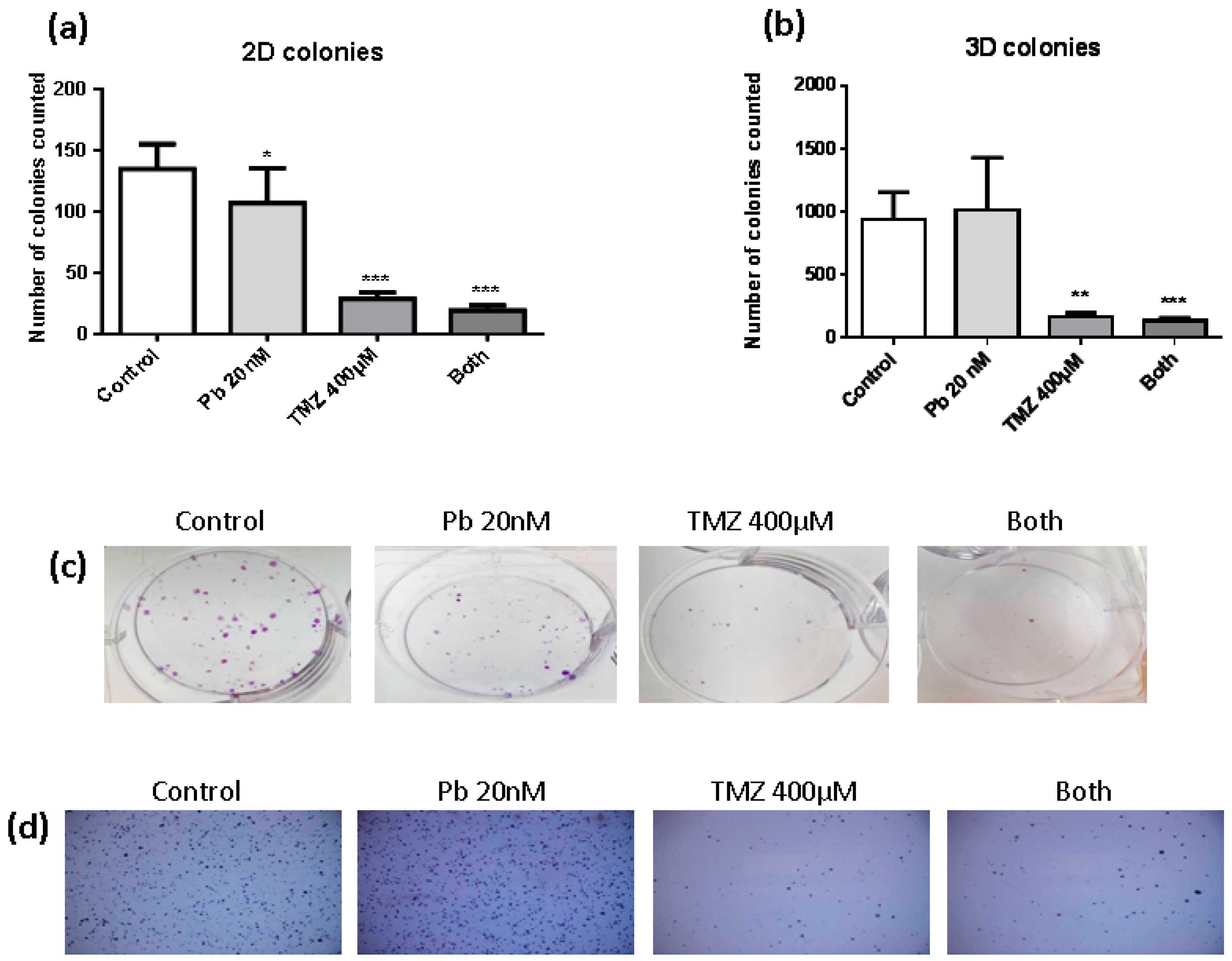
2.1. Temozolomide Inhibits the Clonogenic Capacity of LN405 Glioblastoma Cell Line
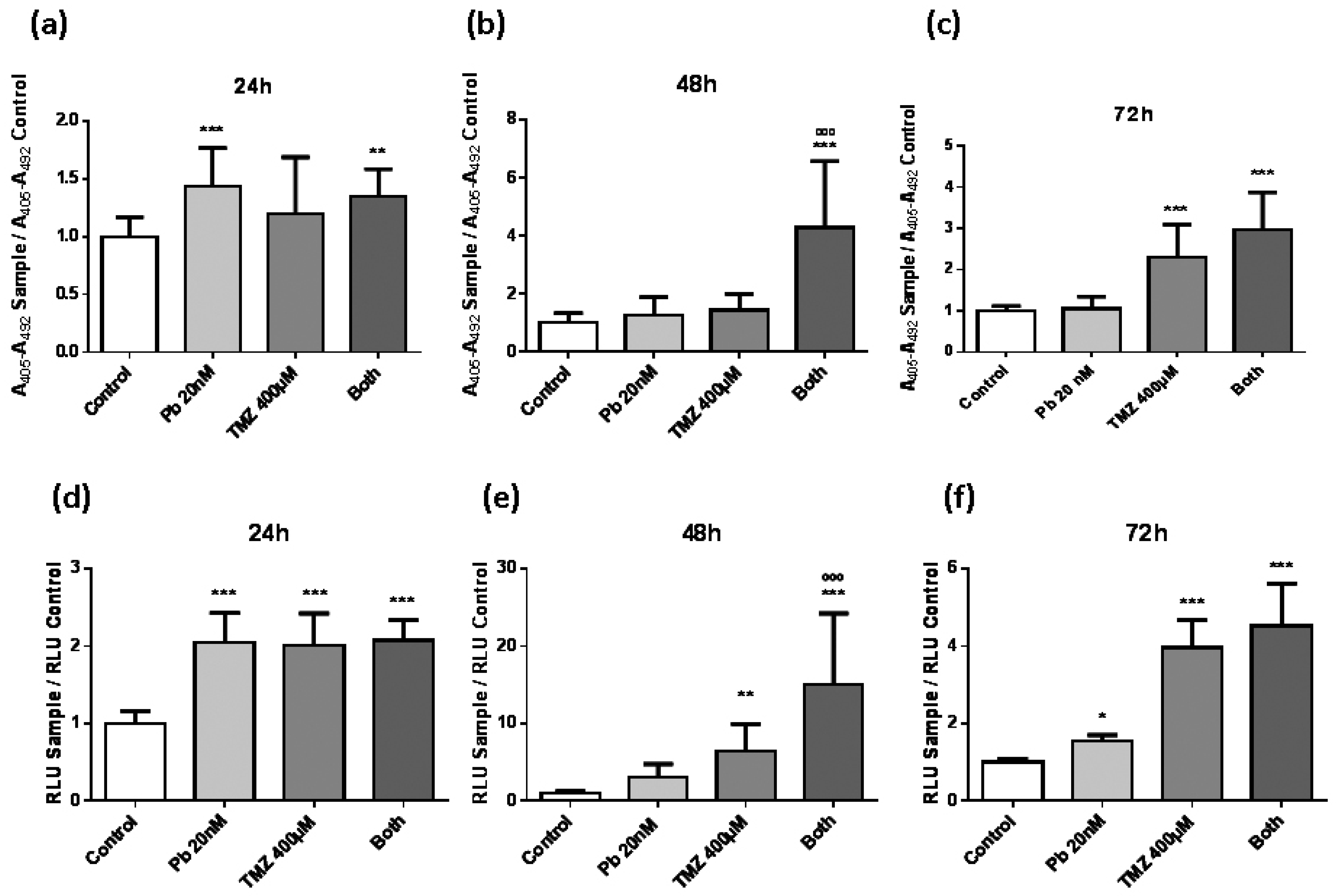
2.2. Panobinostat Accelerates the Effect of Temozolomide to Induce Apoptosis in LN405 Glioblastoma Cells
2.3. Panobinostat Treatment and Its Combination with Temozolomide Increase the Expression of Epithelial Markers and Decrease the Expression of Mesenchymal Markers Related with Epithelial-to-Mesenchymal Transition
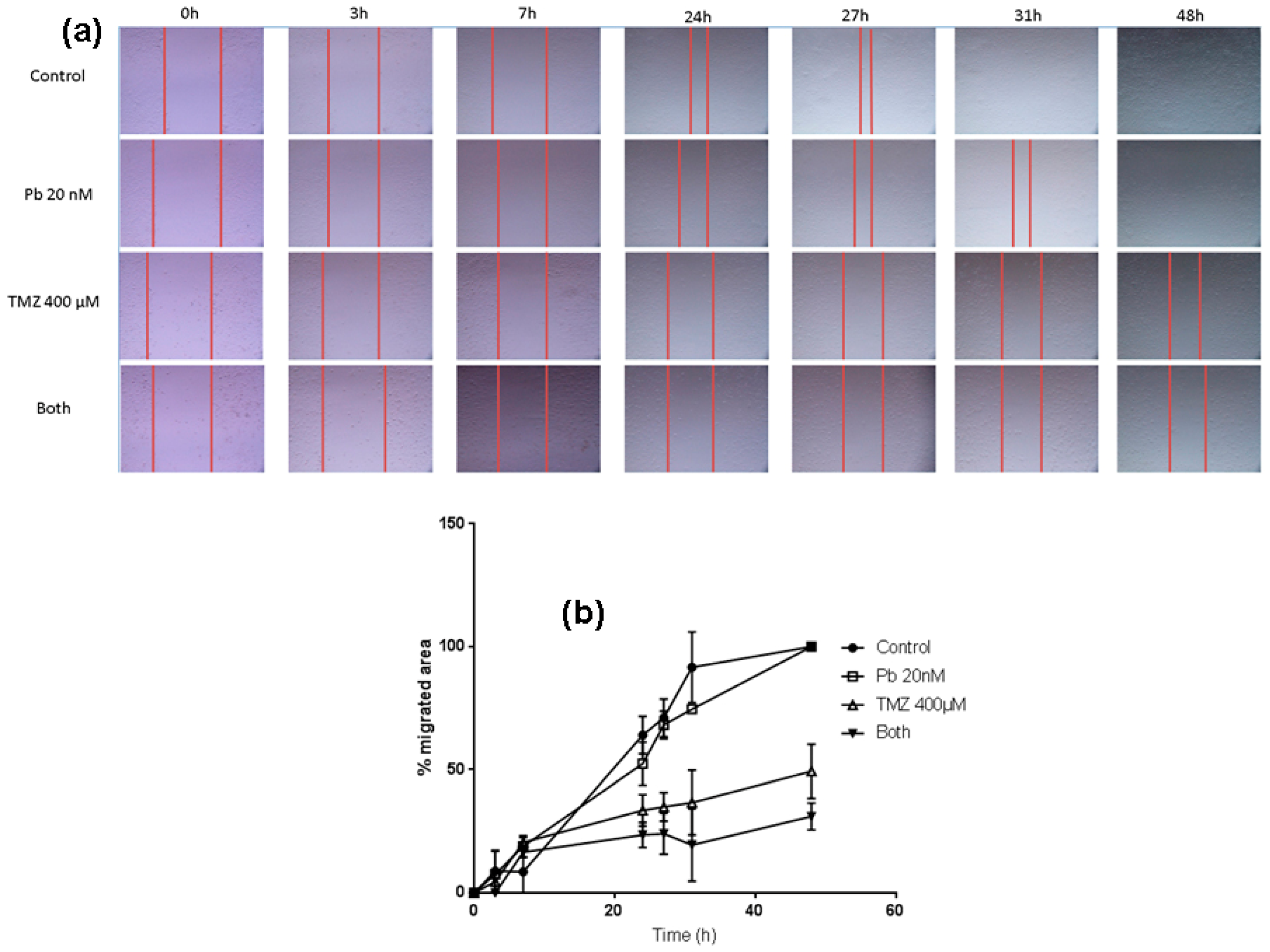
2.4. The Combination of Panobinostat and Temozolomide Reduces Cell Migration with Respect to Temozolomide Alone
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Pharmacologic Treatment
4.3. 2D Colony Formation Assay
4.4. 3D Colony Formation Assay in Soft Agar
4.5. Wound Healing Assay
4.6. Cell Detection Enzyme-Linked Immunosorbent AssayPLUS
4.7. Caspase-Glo 3/7 Assay
4.8. Protein Extraction
4.9. Western Blot
4.10. Analytical Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jovcevska, I.; Kocevar, N.; Komel, R. Glioma and glioblastoma—How much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.A.; Gilbert, M. Targeted therapy in gliomas. Curr. Oncol. Rep. 2014, 16, 379. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W. EGFR-targeted therapy in malignant glioma: Novel aspects and mechanisms of drug resistance. Curr. Mol. Pharmacol. 2010, 3, 37–52. [Google Scholar] [CrossRef]
- Mathews, L.A.; Crea, F.; Farrar, W.L. Epigenetic gene regulation in stem cells and correlation to cancer. Differentiation 2009, 78, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2016, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Kuljaca, S.; Tee, A.; Marshall, G.M. Histone deacetylase inhibitors: Multifunctional anticancer agents. Cancer Treat. Rev. 2006, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lotte, M.E.; Pont, B.; Kleijn, A.; Kloezeman, J.J.; van den Bossche, W.; Kaufmann, J.K.; de Vrij, J.; Leenstra, S.; Clemens, M.F.; Dirven, C.M.; et al. The HDAC inhibitors scriptaid and LBH589 combined with the oncolytic virus delta24-RGD exert enhanced anti-tumor efficacy in patient-derived glioblastoma cells. PLoS ONE 2015, 10, e0127058. [Google Scholar] [CrossRef]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Lotte, M.E.; Pont, B.; Naipal, K.; Kloezeman, J.J.; Venkatesan, S.; van den Bent, M.; van Gent, D.C.; Dirven, C.M.; Kanaar, R.; Lamfers, M.L.; et al. DNA damage response and anti-apoptotic proteins predict radiosensitization efficacy of HDAC inhibitors SAHA and LBH589 in patient-derived glioblastoma cells. Cancer Lett. 2015, 356, 525–535. [Google Scholar]
- Singleton, W.G.; Collins, A.M.; Bienemann, A.S.; Killick-Cole, C.L.; Haynes, H.R.; Asby, D.J.; Butts, C.P.; Wyatt, M.J.; Barua, N.U.; Gill, S.S. Convection enhanced delivery of panobinostat (LBH589)-loaded pluronicnano-micelles prolongs survival in the F98 rat glioma model. Int. J. Nanomed. 2017, 12, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.G.; Li, W.H.; Hua, F.; Cheng, H.X.; Zhao, M.Q.; Sun, X.C.; Qin, Y.J.; Li, J.M. LBH589 inhibits glioblastoma growth and angiogenesis through suppression of HIF-1αexpression. J. Neuropathol. Exp. Neurol. 2017, 76, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Friday, B.B.; Yang, L.; Atadja, P.; Wigle, D.; Sarkaria, J.; Adjei, A.A. Mitochondrial Bax translocation partially mediates synergistic cytotoxicity between histone deacetylase inhibitors and proteasome inhibitors in glioma cells. Neuro Oncol. 2008, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Skuli, N.; Karpel-Massler, G.; Frosina, G.; Ryken, T.; Halatsch, M.E. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: The EIS regimen. Oncotarget 2017, 8, 60727–60749. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Fortunati, N.; Pugliese, M.; Marano, F.; Ortoleva, L.; Poli, R.; Asioli, S.; Bandino, A.; Palestini, N.; Grange, C.; et al. Histone deacetylase inhibition modulates E-cadherin expression and suppresses migration and invasion of anaplastic thyroid cancer cells. J. Clin. Endocrinol. Metab. 2012, 97, E1150–E1159. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Zhang, X.J.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of α-tubulin acetylation is associated with TGF-β -induced epithelial-mesenchymal transition. J. Biol. Chem. 2016, 291, 5396–5405. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Amulticentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Assouline, S.E.; Nielsen, T.H.; Yu, S.; Alcaide, M.; Chong, L.; MacDonald, D.; Tosikyan, A.; Kukreti, V.; Kezouh, A.; Petrogiannis-Haliotis, T.; et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood 2016, 128, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Phipps, C.; Hwang, W.Y.; Tan, S.Y.; Yeap, C.H.; Chan, Y.H.; Tay, K.; Lim, S.T.; Lee, Y.S.; Kumar, S.G.; et al. Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: An open-label, multicentre phase 2 trial. Lancet Haematol. 2015, 2, e326–e333. [Google Scholar] [CrossRef]
- De Marinis, F.; Atmaca, A.; Tiseo, M.; Giuffreda, L.; Rossi, A.; Gebbia, V.; D’Antonio, C.; DalZotto, L.; Al-Batran, S.E.; Marsoni, S.; et al. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Investig. 2011, 29, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Lubner, S.J.; Mulkerin, D.L.; Rajguru, S.; Carmichael, L.; Chen, H.; Holen, K.D.; LoConte, N.K. A phase II trial of a histone deacetylase inhibitor panobinostat in patients with low-grade neuroendocrine tumors. Oncologist 2016, 21, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Hilger, R.A.; Muhlenberg, T.; Grabellus, F.; Nagarajah, J.; Hoiczyk, M.; Reichardt, A.; Ahrens, M.; Reichardt, P.; Grunewald, S.; et al. Phase I study of panobinostat and imatinib in patients with treatment-refractory metastatic gastrointestinal stromal tumors. Br. J. Cancer 2014, 110, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, A.; Hatake, K.; Matsui, K.; Sakajiri, S.; Hirashima, T.; Tanii, H.; Kobayashi, K.; Yamamoto, N. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Haura, E.; Chiappori, A.; Tanvetyanon, T.; Williams, C.C.; Pinder-Schenck, M.; Kish, J.A.; Kreahling, J.; Lush, R.; Neuger, A.; et al. A phase I, pharmacokinetic, and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin. Cancer Res. 2014, 20, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Buchbinder, E.I.; Granter, S.R.; Rodig, S.J.; Giobbie-Hurder, A.; Becerra, C.; Tsiaras, A.; Gjini, E.; Fisher, D.E.; Hodi, F.S. A phase I trial of panobinostat (LBH589) in patients with metastatic melanoma. Cancer Med. 2016, 5, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Bendell, J.C.; Infante, J.R.; Spigel, D.R.; Thompson, D.S.; Yardley, D.A.; Greco, F.A.; Murphy, P.B.; Burris, H.A. 3rd. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin. Adv. Hematol. Oncol. 2011, 9, 225–230. [Google Scholar] [PubMed]
- Jones, S.F.; Infante, J.R.; Thompson, D.S.; Mohyuddin, A.; Bendell, J.C.; Yardley, D.A.; Burris, H.A., 3rd. A phase I trial of oral administration of panobinostat in combination with paclitaxel and carboplatin in patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 471–475. [Google Scholar]
- Morita, S.; Oizumi, S.; Minami, H.; Kitagawa, K.; Komatsu, Y.; Fujiwara, Y.; Inada, M.; Yuki, S.; Kiyota, N.; Mitsuma, A.; et al. Phase I dose-escalating study of panobinostat (LBH589) administered intravenously to Japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Witteveen, P.O.; Lolkema, M.P.; Hess, D.; Gelderblom, H.; Hussain, S.A.; Porro, M.G.; Waldron, E.; Valera, S.Z.; Mu, S. A phase I, open-label, multicenter study to evaluate the pharmacokinetics and safety of oral panobinostat in patients with advanced solid tumors and varying degrees of renal function. Cancer Chemother. Pharmacol. 2015, 75, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Starodub, A.N.; Jia, J.; Meadows, K.L.; Nixon, A.B.; Dellinger, A.; Morse, M.A.; Uronis, H.E.; Marcom, P.K.; Zafar, S.Y.; et al. Phase I study of bevacizumab, everolimus, and panobinostat (LBH-589) in advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Takhar, H.S.; Singhal, N.; Gowda, R.; Penniment, M.; Takhar, P.; Brown, M.P. Phase I study evaluating the safety and efficacy of oral panobinostat in combination with radiotherapy or chemoradiotherapy in patients with inoperable stage III non-small-cell lung cancer. Anticancer Drugs 2015, 26, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I study of panobinostat(LBH589) and letrozole in postmenopausal metastatic breast cancer patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Zahoor, H.; McLaughlin, B.; Gooding, W.E.; Schmitz, J.C.; Siegfried, J.M.; Socinski, M.A.; Argiris, A. Phase I trial of carboplatin and etoposide in combination with panobinostat in patients with lung cancer. Anticancer Res. 2013, 33, 4475–4481. [Google Scholar] [PubMed]
- Thomas, S.; Aggarwal, R.; Jahan, T.; Ryan, C.; Troung, T.; Cripps, A.M.; Raha, P.; Thurn, K.T.; Chen, S.; Grabowsky, J.A.; et al. A phase I trial of panobinostat and epirubicin in solid tumors with a dose expansion in patients with sarcoma. Ann. Oncol. 2016, 27, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Drappatz, J.; Lee, E.Q.; Hammond, S.; Grimm, S.A.; Norden, A.D.; Beroukhim, R.; Gerard, M.; Schiff, D.; Chi, A.S.; Batchelor, T.T.; et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J. Neurooncol. 2012, 107, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Palmer, J.D.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Bar-Ad, V.; Judy, K.; Farrell, C.; et al. Phase I trial of panobinostat and fractionated stereotactic re-irradiation therapy for recurrent high-grade gliomas. J. Neurooncol. 2016, 127, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef]
- GraphPath software. Available online: https://www.graphpad.com/scientific-software/prism/ (accessed on 15 November 2017).




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urdiciain, A.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Panobinostat Potentiates Temozolomide Effects and Reverses Epithelial–Mesenchymal Transition in Glioblastoma Cells. Epigenomes 2018, 2, 5. https://doi.org/10.3390/epigenomes2010005
Urdiciain A, Meléndez B, Rey JA, Idoate MA, Castresana JS. Panobinostat Potentiates Temozolomide Effects and Reverses Epithelial–Mesenchymal Transition in Glioblastoma Cells. Epigenomes. 2018; 2(1):5. https://doi.org/10.3390/epigenomes2010005
Chicago/Turabian StyleUrdiciain, Alejandro, Bárbara Meléndez, Juan A. Rey, Miguel A. Idoate, and Javier S. Castresana. 2018. "Panobinostat Potentiates Temozolomide Effects and Reverses Epithelial–Mesenchymal Transition in Glioblastoma Cells" Epigenomes 2, no. 1: 5. https://doi.org/10.3390/epigenomes2010005
APA StyleUrdiciain, A., Meléndez, B., Rey, J. A., Idoate, M. A., & Castresana, J. S. (2018). Panobinostat Potentiates Temozolomide Effects and Reverses Epithelial–Mesenchymal Transition in Glioblastoma Cells. Epigenomes, 2(1), 5. https://doi.org/10.3390/epigenomes2010005