
Increase of DNA Methylation at the HvCKX2.1 Promoter by Terminal Drought Stress in Barley

 , and
, and
Abstract
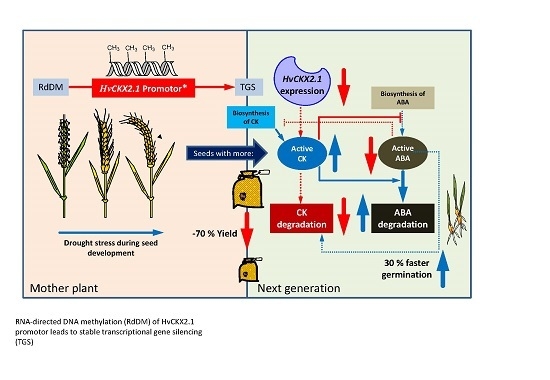
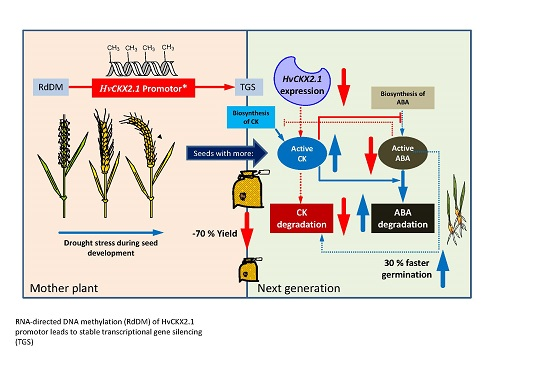
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Identification of Terminal Drought Stress Specific Small RNAs in Barley Caryopses
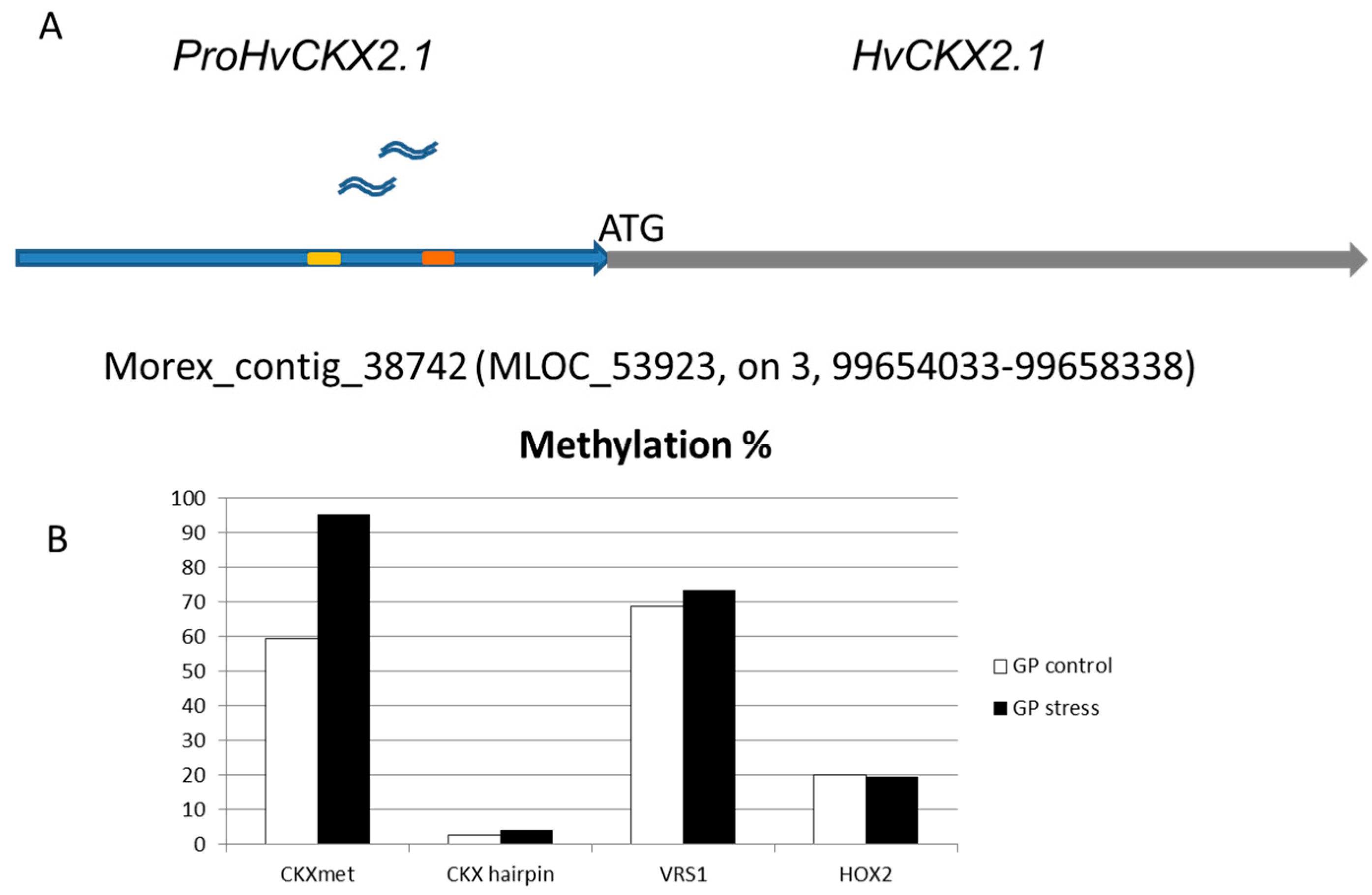
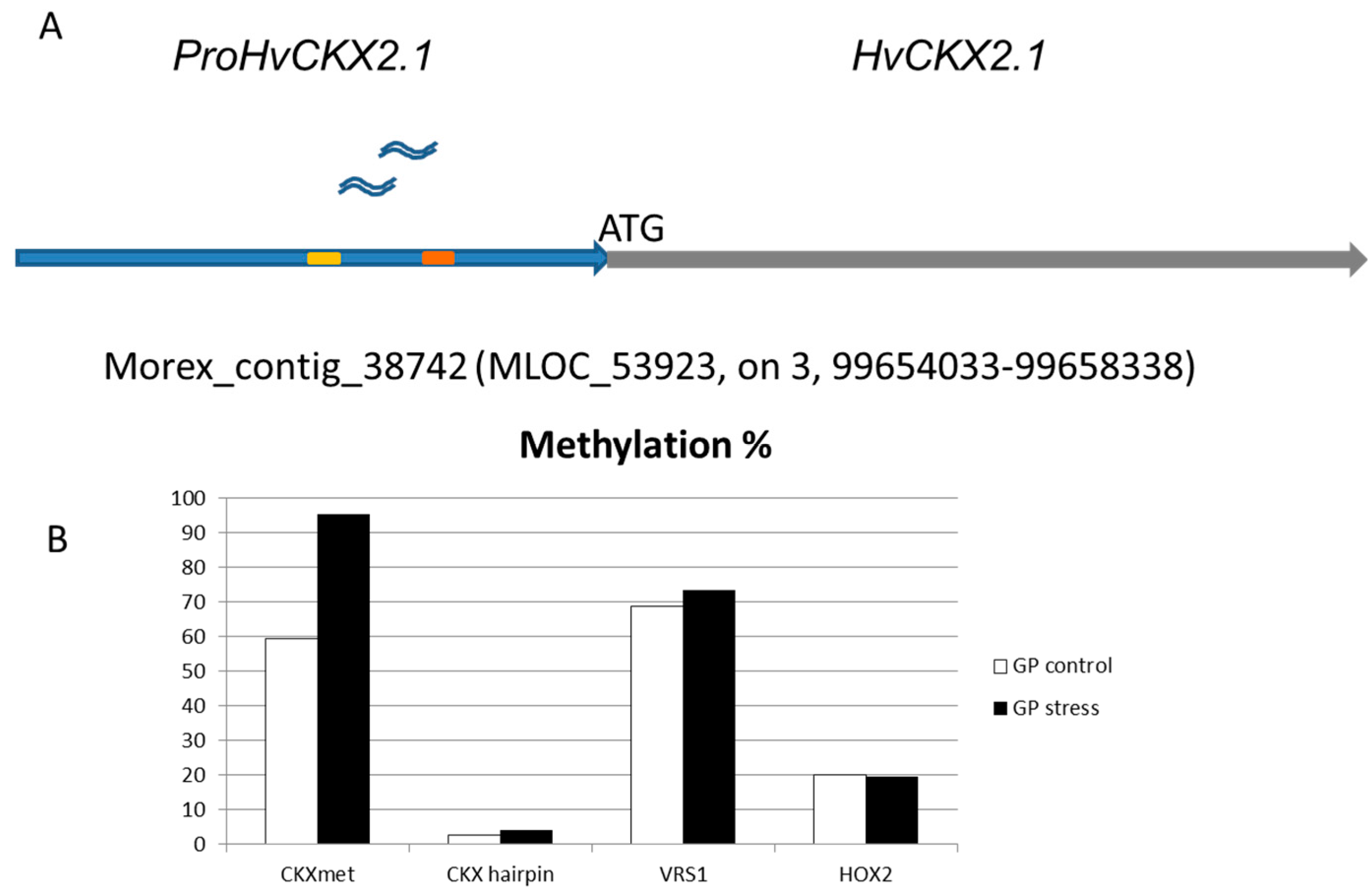
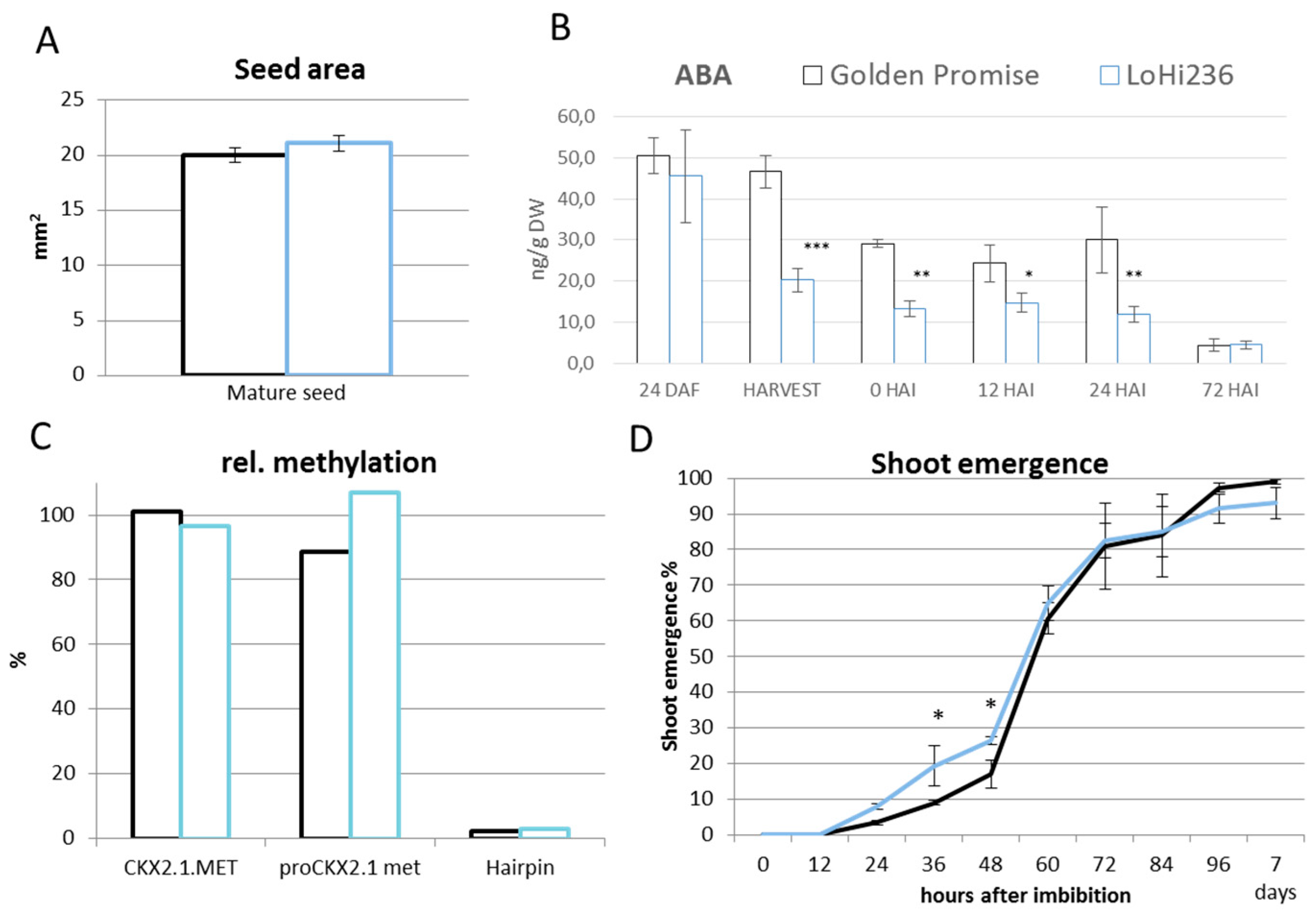
2.2. Methylation Status of ProHvCKX2.1
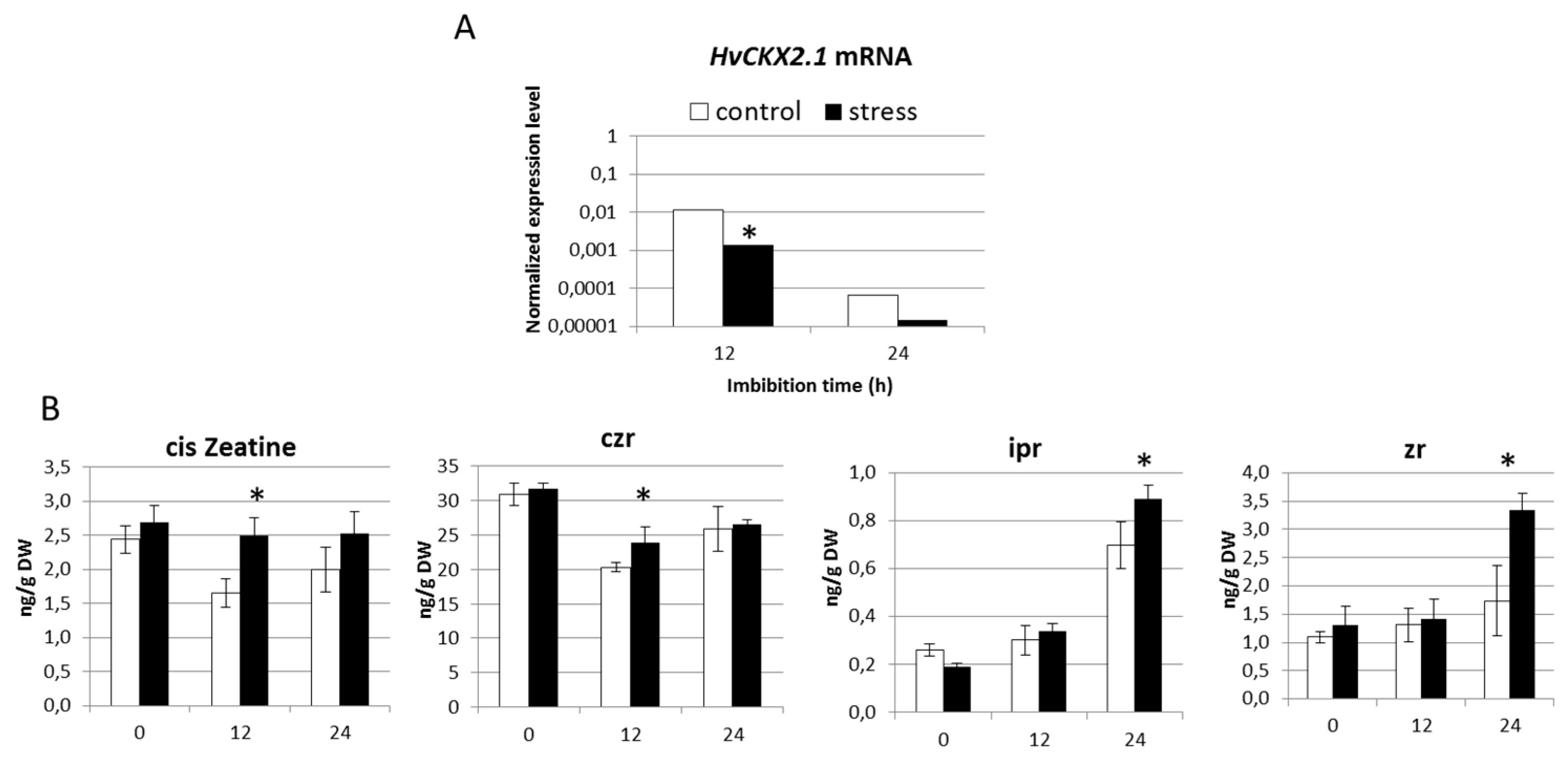
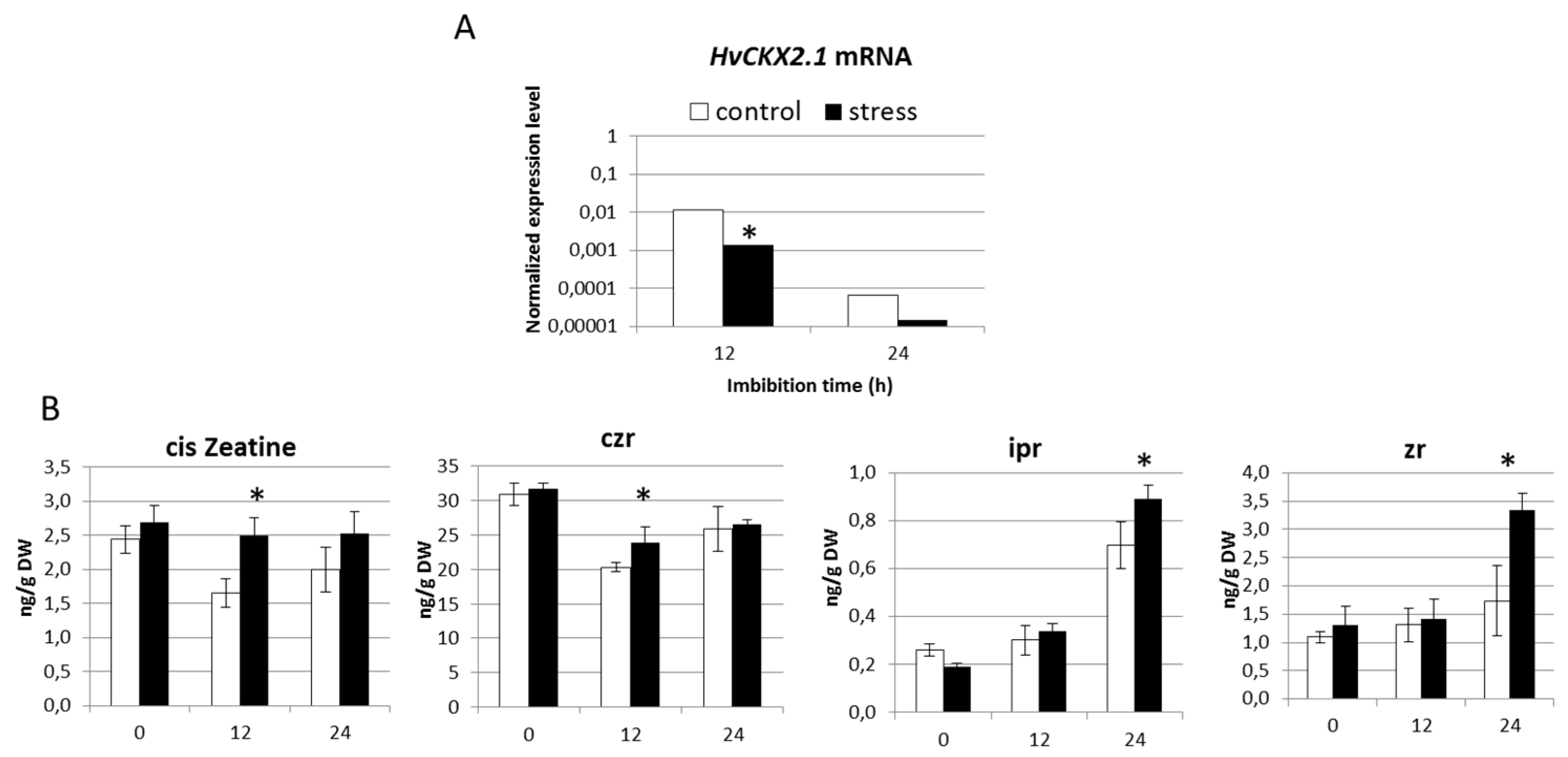
2.3. Seedlings Derived from Drought Stress Plants Exhibit Reduced Presence of HvCKX2.1 mRNA
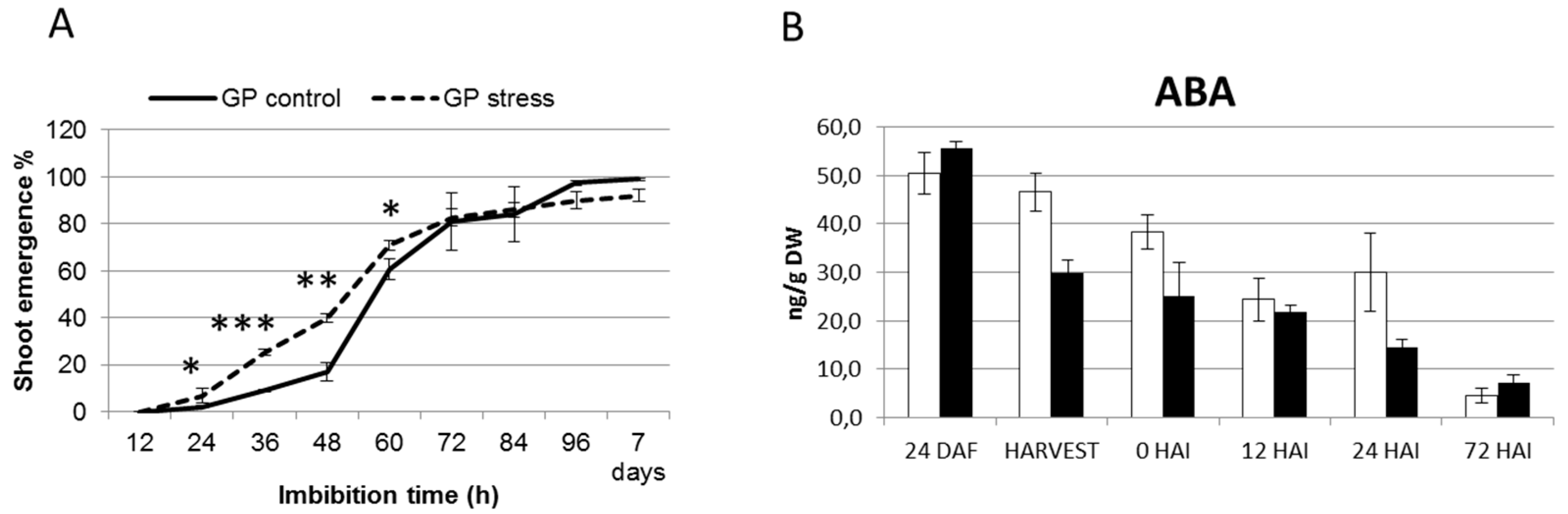
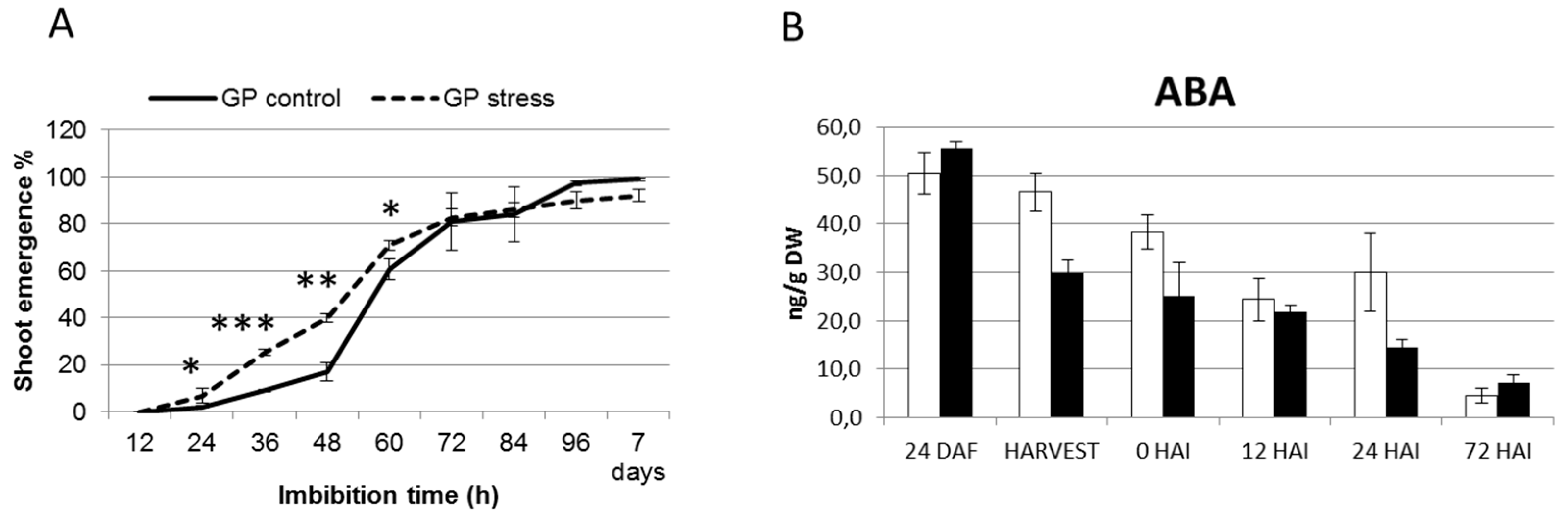
2.4. Terminal Drought Stress Enhances the Shoot Emergence Rate of Seeds
2.5. Barley with Increased Levels of ABA During Grain Development Phase Show Faster Germination
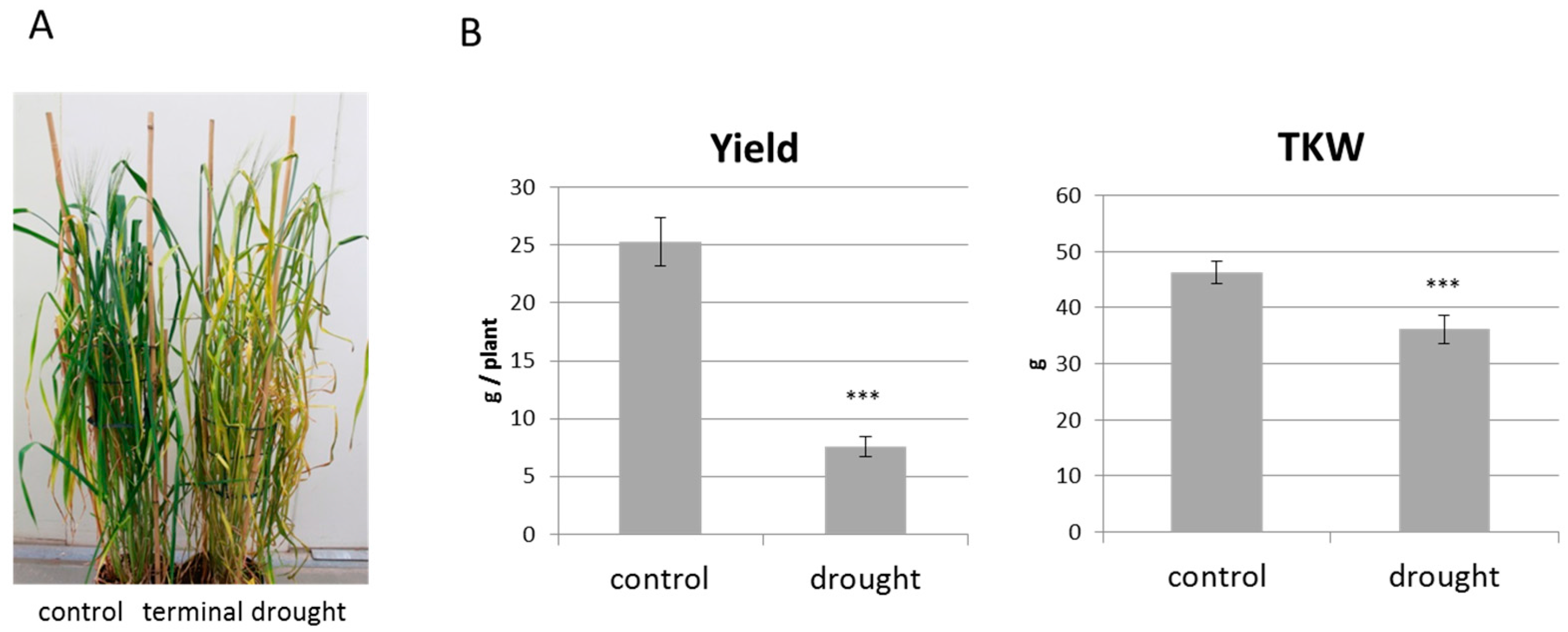
3. Discussion
4. Material and Methods
4.1. Plant Cultivation and Used Barley Plants
4.2. Small RNA Sequencing
- “hairpin_and_mature_Hordeum_vulgare.fa”from miRBASE
- “hairpin_and_mature.fa”from miRBASE
- “nt.viridiplantae.fas”
4.3. Transcript Quantification
4.4. DNA Methylation Analysis
4.5. Plant Hormone (PH) Analysis -Abscisic Acid, Cytokinin, Auxin and Salicylic Acid Extraction
Accession Numbers
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Loss, S.P.; Siddique, K.H.M. Morphological and physiological traits associated with wheat yield increases in mediterranean environments. Adv. Agron. 1994, 52, 229–276. [Google Scholar]
- Fereres, E.; Orgaz, F.; Gonzalez-Dugo, V. Reflections on food security under water scarcity. J. Exp. Bot. 2011, 62, 4079–4086. [Google Scholar] [CrossRef] [PubMed]
- Overpeck, J.; Udall, B. Dry times ahead. Science 2010, 328, 1642–1643. [Google Scholar] [CrossRef] [PubMed]
- Govind, G.; Seiler, C.; Wobus, U.; Sreenivasulu, N. Importance of aba homeostasis under terminal drought stress in regulating grain filling events. Plant Signal Behav. 2011, 6, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Harshavardhan, V.T.; Govind, G.; Seiler, C.; Kohli, A. Contrapuntal role of aba: Does it mediate stress tolerance or plant growth retardation under long-term drought stress? Gene 2012, 506, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Radchuk, V.V.; Sreenivasulu, N.; Radchuk, R.I.; Wobus, U.; Weschke, W. The methylation cycle and its possible functions in barley endosperm development. Plant Mol. Biol. 2005, 59, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Wolff, P.; Spillane, C. Epigenetic mechanisms underlying genomic imprinting in plants. Annu. Rev. Plant Biol. 2012, 63, 331–352. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Kanno, T.; Matzke, A.J. Rna-directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.L.; Li, Z.H. The functions of micrornas in plants. Front. Biosci. 2007, 12, 3975–3982. [Google Scholar] [PubMed]
- Rubio-Somoza, I.; Weigel, D. Microrna networks and developmental plasticity in plants. Trends Plant Sci. 2011, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ning, S.; Wu, J.; Tagiri, A.; Komatsuda, T. An epiallele at cly1 affects the expression of floret closing (cleistogamy) in barley. Genetics 2015, 199, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Houston, K.; McKim, S.M.; Comadran, J.; Bonar, N.; Druka, I.; Uzrek, N.; Cirillo, E.; Guzy-Wrobelska, J.; Collins, N.C.; Halpin, C.; et al. Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc. Natl. Acad. Sci. USA 2013, 110, 16675–16680. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, J.; Hussain, S.S.; Shi, B.J. Role of micrornas in plant drought tolerance. Plant Biotechnol. J 2015, 13, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Alba, A.E.; Elvira-Matelot, E.; Vaucheret, H. Gene silencing in plants: A diversity of pathways. Biochim. Biophys. Acta 2013, 1829, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.; Vaucheret, H. Form, function, and regulation of argonaute proteins. Plant Cell 2010, 22, 3879–3889. [Google Scholar] [CrossRef] [PubMed]
- He, X.-J.; Chen, T.; Zhu, J.-K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011, 21, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.; Kanno, T.; Huettel, B.; Daxinger, L.; Matzke, A.J.M. Targets of rna-directed DNA methylation. Curr. Opin. Plant Biol. 2007, 10, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Kalladan, R.; Worch, S.; Rolletschek, H.; Harshavardhan, V.T.; Kuntze, L.; Seiler, C.; Sreenivasulu, N.; Roder, M.S. Identification of quantitative trait loci contributing to yield and seed quality parameters under terminal drought in barley advanced backcross lines. Mol. Breed. 2013, 32, 71–90. [Google Scholar] [CrossRef]
- Seiler, C.; Harshavardhan, V.T.; Reddy, P.S.; Hensel, G.; Kumlehn, J.; Eschen-Lippold, L.; Rajesh, K.; Korzun, V.; Wobus, U.; Lee, J.; et al. Abscisic acid flux alterations result in differential aba signalling responses and impact assimilation efficiency in barley under terminal drought stress. Plant Physiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Muller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA ends (mace) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, M. DNA methylation of the CKX2.1 promotor by terminal drought stress in barley. e!DAL 2017. [Google Scholar] [CrossRef]
- Arend, D.; Junker, A.; Scholz, U.; Schuler, D.; Wylie, J.; Lange, M. PGP repository: A plant phenomics and genomics data publication infrastructure. Database (Oxford) 2016, 2016. [Google Scholar]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [PubMed]
- Zhai, J.; Liu, J.; Liu, B.; Li, P.; Meyers, B.C.; Chen, X.; Cao, X. Small RNA-directed epigenetic natural variation in arabidopsis thaliana. PLoS Genet. 2008, 4, e1000056. [Google Scholar]
- Mascher, M.; Muehlbauer, G.J.; Rokhsar, D.S.; Chapman, J.; Schmutz, J.; Barry, K.; Munoz-Amatriain, M.; Close, T.J.; Wise, R.P.; Schulman, A.H.; et al. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J. 2013, 76, 718–727. [Google Scholar] [PubMed]
- McGaw, B.A.; Horgan, R. Cytokinin oxidase from Zea mays kernels Andvinca rosea crown-gall tissue. Planta 1983, 159, 30–37. [Google Scholar] [PubMed]
- Mameaux, S.; Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Jack, P.; Werner, P.; Gray, J.C.; Greenland, A.J.; et al. Molecular, phylogenetic and comparative genomic analysis of the cytokinin oxidase/dehydrogenase gene family in the Poaceae. Plant Biotechnol. J. 2012, 10, 67–82. [Google Scholar] [PubMed]
- Zalewski, W.; Gasparis, S.; Boczkowska, M.; Rajchel, I.K.; Kala, M.; Orczyk, W.; Nadolska-Orczyk, A. Expression patterns of HvCKX genes indicate their role in growth and reproductive development of barley. PLoS ONE 2014, 9, e115729. [Google Scholar]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The GYPSY database (GYDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [PubMed]
- Chang, T.H.; Huang, H.Y.; Hsu, J.B.K.; Weng, S.L.; Horng, J.T.; Huang, H.D. An enhanced computational platform for investigating the roles of regulatory rna and for identifying functional RNA motifs. BMC Bioinform. 2013, 14. [Google Scholar]
- Zhang, H.; He, X.; Zhu, J.K. RNA-directed DNA methylation in plants: Where to start? RNA Biol. 2013, 10, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Galuszka, P.; Frebortova, J.; Werner, T.; Yamada, M.; Strnad, M.; Schmulling, T.; Frebort, I. Cytokinin oxidase/dehydrogenase genes in barley and wheat: Cloning and heterologous expression. Eur. J. Biochem. 2004, 271, 3990–4002. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, W.; Galuszka, P.; Gasparis, S.; Orczyk, W.; Nadolska-Orczyk, A. Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J. Exp. Bot. 2010, 61, 1839–1851. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, W.; Orczyk, W.; Gasparis, S.; Nadolska-Orczyk, A. HvCKX2 gene silencing by biolistic or agrobacterium-mediated transformation in barley leads to different phenotypes. BMC Plant Biol. 2012, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Mrizova, K.; Jiskrova, E.; Vyroubalova, S.; Novak, O.; Ohnoutkova, L.; Pospisilova, H.; Frebort, I.; Harwood, W.A.; Galuszka, P. Overexpression of cytokinin dehydrogenase genes in barley (Hordeum vulgare cv. Golden promise) fundamentally affects morphology and fertility. PLoS ONE 2013, 8, e79029. [Google Scholar] [CrossRef] [PubMed]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Fullgrabe, A.; Fuentes, A.M.; Jupp, S.; Koskinen, S.; et al. Expression atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.X.; Waugh, R.; Langridge, P.; Close, T.J.; Wise, R.P.; Graner, A.; Matsumoto, T.; Sato, K.; Schulman, A.; Muehlbauer, G.J.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [PubMed]
- Malito, E.; Coda, A.; Bilyeu, K.D.; Fraaije, M.W.; Mattevi, A. Structures of Michaelis and product complexes of plant cytokinin dehydrogenase: Implications for flavoenzyme catalysis. J. Mol. Biol. 2004, 341, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, H. Cytokinins: Activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Graeber, K.; Nakabayashi, K.; Miatton, E.; Leubner-Metzger, G.; Soppe, W.J. Molecular mechanisms of seed dormancy. Plant Cell Environ. 2012, 35, 1769–1786. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Ye, T.; Zhao, S.; Liu, Z.; Feng, Y.Q.; Wu, Y. Cytokinin antagonizes ABA suppression to seed germination of arabidopsis by downregulating abi5 expression. Plant J. 2011, 68, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Harshavardhan, V.T.; Rajesh, K.; Reddy, P.S.; Strickert, M.; Rolletschek, H.; Scholz, U.; Wobus, U.; Sreenivasulu, N. ABA biosynthesis and degradation contributing to ABA homeostasis during barley seed development under control and terminal drought-stress conditions. J. Exp. Bot. 2011, 62, 2615–2632. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, R.; Bhalothia, P.; Bansal, P.; Basantani, M.K.; Bharti, V.; Mehrotra, S. Abscisic acid and abiotic stress tolerance—Different tiers of regulation. J. Plant Physiol. 2014, 171, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, K.; Kapazoglou, A.; Tondelli, A.; Francia, E.; Stanca, M.A.; Bladenopoulos, K.; Tsaftaris, A.S. Epigenetic chromatin modifiers in barley: I. Cloning, mapping and expression analysis of the plant specific HD2 family of histone deacetylases from barley, during seed development and after hormonal treatment. Physiol. Plant. 2009, 136, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Kapazoglou, A.; Tondelli, A.; Papaefthimiou, D.; Ampatzidou, H.; Francia, E.; Stanca, M.A.; Bladenopoulos, K.; Tsaftaris, A.S. Epigenetic chromatin modifiers in barley: IV. The study of barley polycomb group (PCG) genes during seed development and in response to external ABA. BMC Plant Biol. 2010, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kim, J.M.; Matsunaga, W.; Saze, H.; Matsui, A.; Endo, T.A.; Harukawa, Y.; Takagi, H.; Yaegashi, H.; Masuta, Y.; et al. A stress-activated transposon in arabidopsis induces transgenerational abscisic acid insensitivity. Sci. Rep. 2016, 6, 23181. [Google Scholar] [CrossRef] [PubMed]
- Lewsey, M.G.; Hardcastle, T.J.; Melnyk, C.W.; Molnar, A.; Valli, A.; Urich, M.A.; Nery, J.R.; Baulcombe, D.C.; Ecker, J.R. Mobile small rnas regulate genome-wide DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, E801–E810. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gent, J.I.; Zynda, G.; Song, J.; Makarevitch, I.; Hirsch, C.D.; Hirsch, C.N.; Dawe, R.K.; Madzima, T.F.; McGinnis, K.M.; et al. RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc. Natl. Acad. Sci. USA 2015, 112, 14728–14733. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, A.; Sun, W.; Zhang, J.; Wu, X.; Mousavi, M.; Mohammadi, K.; Yin, T.; Zhuge, Q. RNA-directed DNA methylation in plants. Plant Cell Rep. 2015, 34, 1857–1862. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, S.; Ye, L.; Hu, H.; Li, C.; Zhang, G. The effects of GA and ABA treatments on metabolite profile of germinating barley. Food Chem. 2016, 192, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, J.; Robayo-Romero, M.E.; Gendreau, E.; Benech-Arnold, R.L.; Corbineau, F. Involvement of ABA in induction of secondary dormancy in barley (Hordeum vulgare L.) seeds. Plant Cell Physiol. 2008, 49, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Bokszczanin, K.L.; Krezdorn, N.; Fragkostefanakis, S.; Muller, S.; Rycak, L.; Chen, Y.; Hoffmeier, K.; Kreutz, J.; Paupiere, M.J.; Chaturvedi, P.; et al. Identification of novel small ncRNAs in pollen of tomato. BMC Genom. 2015, 16, 714. [Google Scholar] [CrossRef] [PubMed]
- Finke, A.; Kuhlmann, M.; Mette, M.F. Idn2 has a role downstream of sirna formation in rna-directed DNA methylation. Epigenetics 2012, 7, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Kamada-Nobusada, T.; Komatsu, H.; Takei, K.; Kuroha, T.; Mizutani, M.; Ashikari, M.; Ueguchi-Tanaka, M.; Matsuoka, M.; Suzuki, K.; et al. Highly sensitive and high-throughput analysis of plant hormones using MS-probe modification and liquid chromatography-tandem mass spectrometry: An application for hormone profiling in Oryza sativa. Plant Cell Physiol. 2009, 50, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Jikumaru, Y.; Kamiya, Y. Profiling of hormones and related metabolites in seed dormancy and germination studies. Methods Mol. Biol. 2011, 773, 99–111. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surdonja, K.; Eggert, K.; Hajirezaei, M.-R.; Harshavardhan, V.T.; Seiler, C.; Von Wirén, N.; Sreenivasulu, N.; Kuhlmann, M. Increase of DNA Methylation at the HvCKX2.1 Promoter by Terminal Drought Stress in Barley. Epigenomes 2017, 1, 9. https://doi.org/10.3390/epigenomes1020009
Surdonja K, Eggert K, Hajirezaei M-R, Harshavardhan VT, Seiler C, Von Wirén N, Sreenivasulu N, Kuhlmann M. Increase of DNA Methylation at the HvCKX2.1 Promoter by Terminal Drought Stress in Barley. Epigenomes. 2017; 1(2):9. https://doi.org/10.3390/epigenomes1020009
Chicago/Turabian StyleSurdonja, Korana, Kai Eggert, Mohammad-Reza Hajirezaei, Vokkaliga Thammegowda Harshavardhan, Christiane Seiler, Nicolaus Von Wirén, Nese Sreenivasulu, and Markus Kuhlmann. 2017. "Increase of DNA Methylation at the HvCKX2.1 Promoter by Terminal Drought Stress in Barley" Epigenomes 1, no. 2: 9. https://doi.org/10.3390/epigenomes1020009
APA StyleSurdonja, K., Eggert, K., Hajirezaei, M.-R., Harshavardhan, V. T., Seiler, C., Von Wirén, N., Sreenivasulu, N., & Kuhlmann, M. (2017). Increase of DNA Methylation at the HvCKX2.1 Promoter by Terminal Drought Stress in Barley. Epigenomes, 1(2), 9. https://doi.org/10.3390/epigenomes1020009