
Dynamics of H3K4me3 Chromatin Marks Prevails over H3K27me3 for Gene Regulation during Flower Morphogenesis in Arabidopsis thaliana

 , and
, and 
Abstract
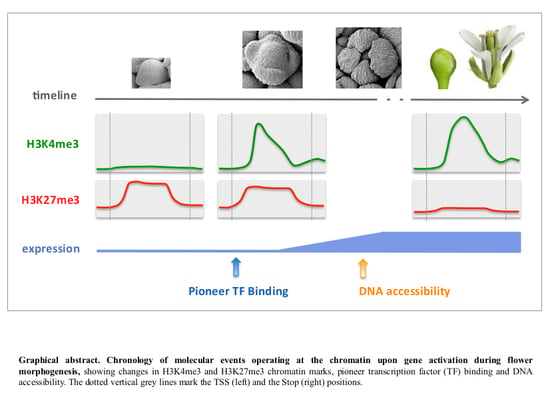
:
1. Introduction
2. Results
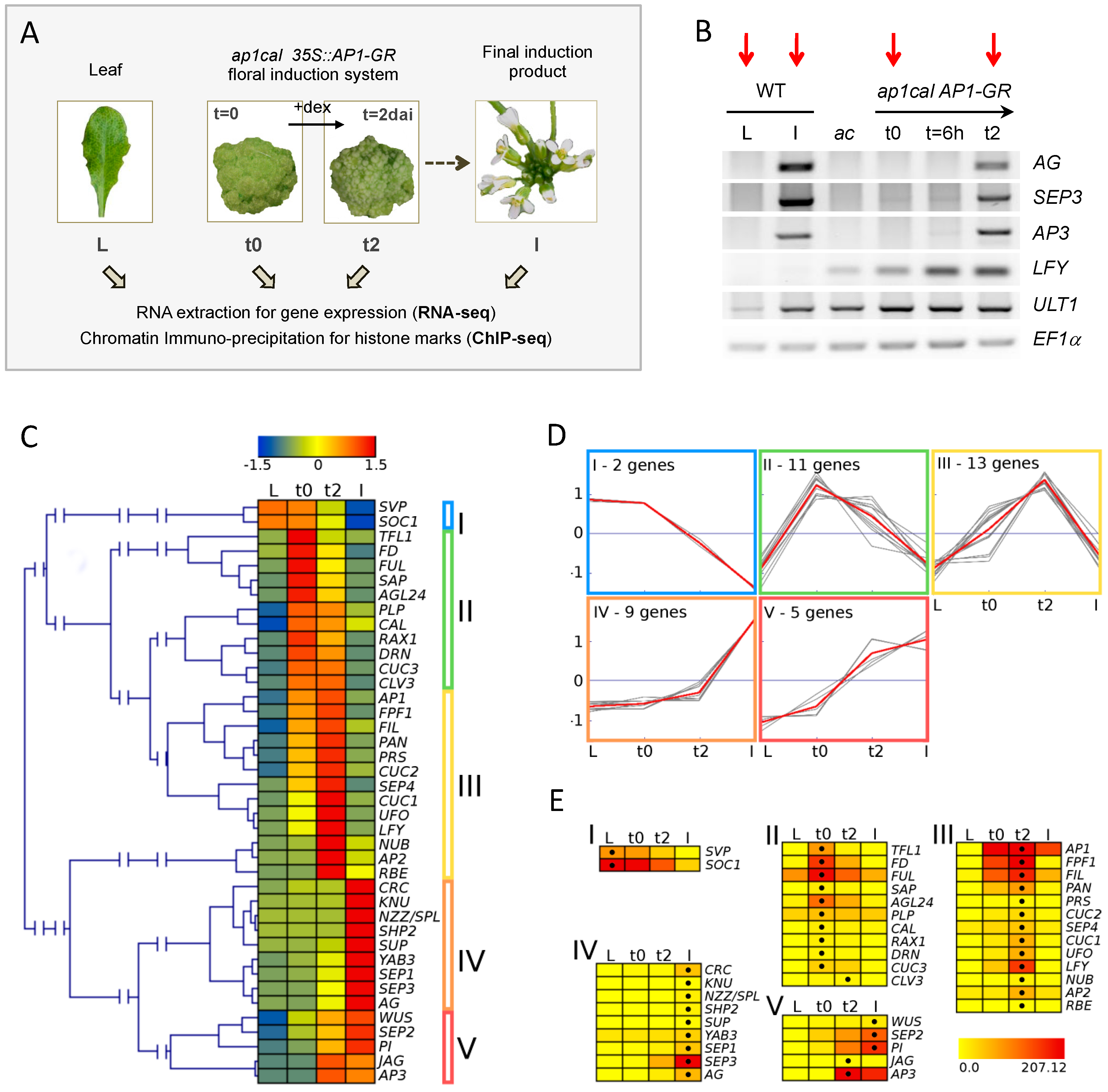
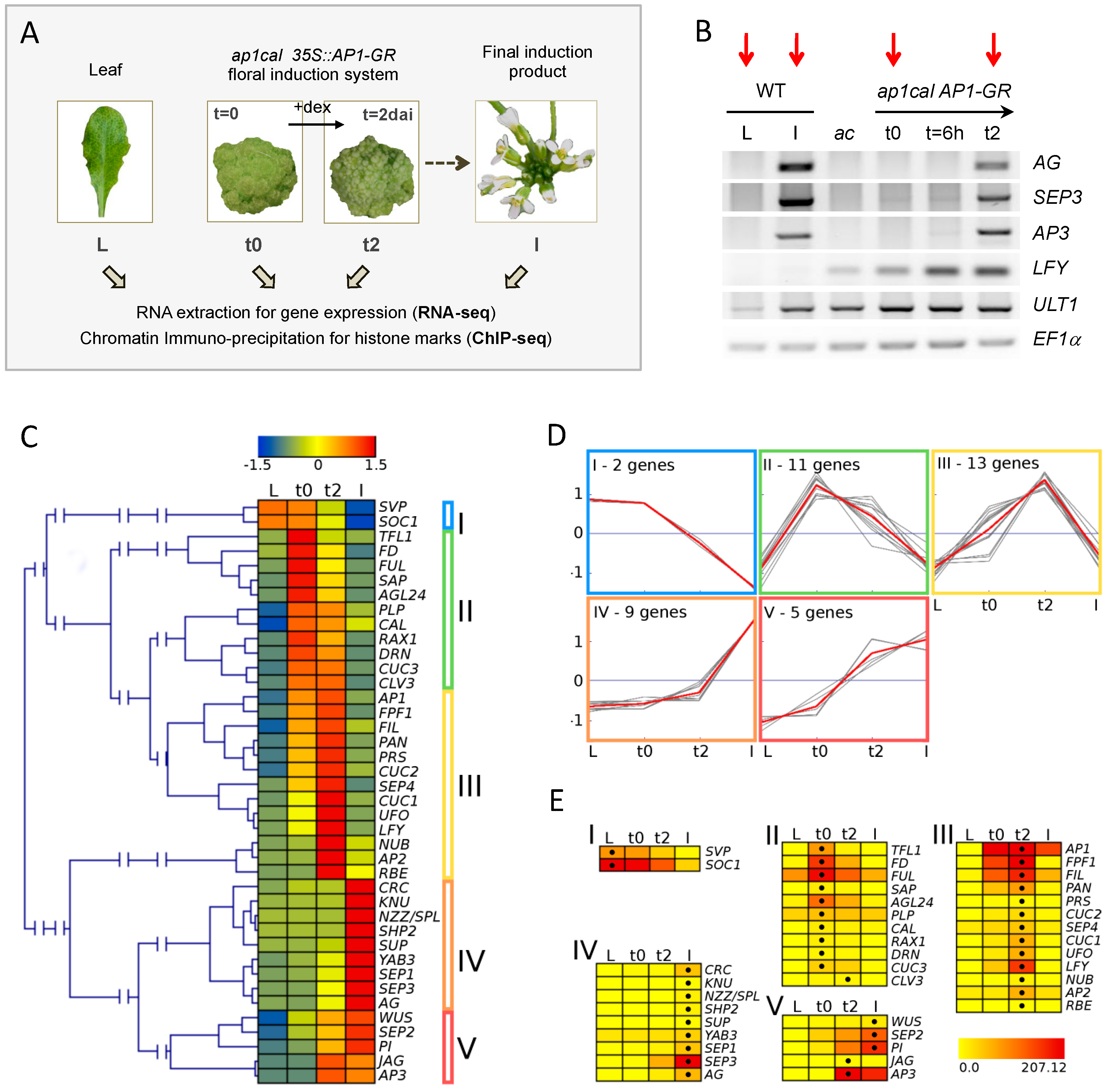
2.1. RNA-seq Reveals Specific Changes in Gene Expression during the Developmental Time Series
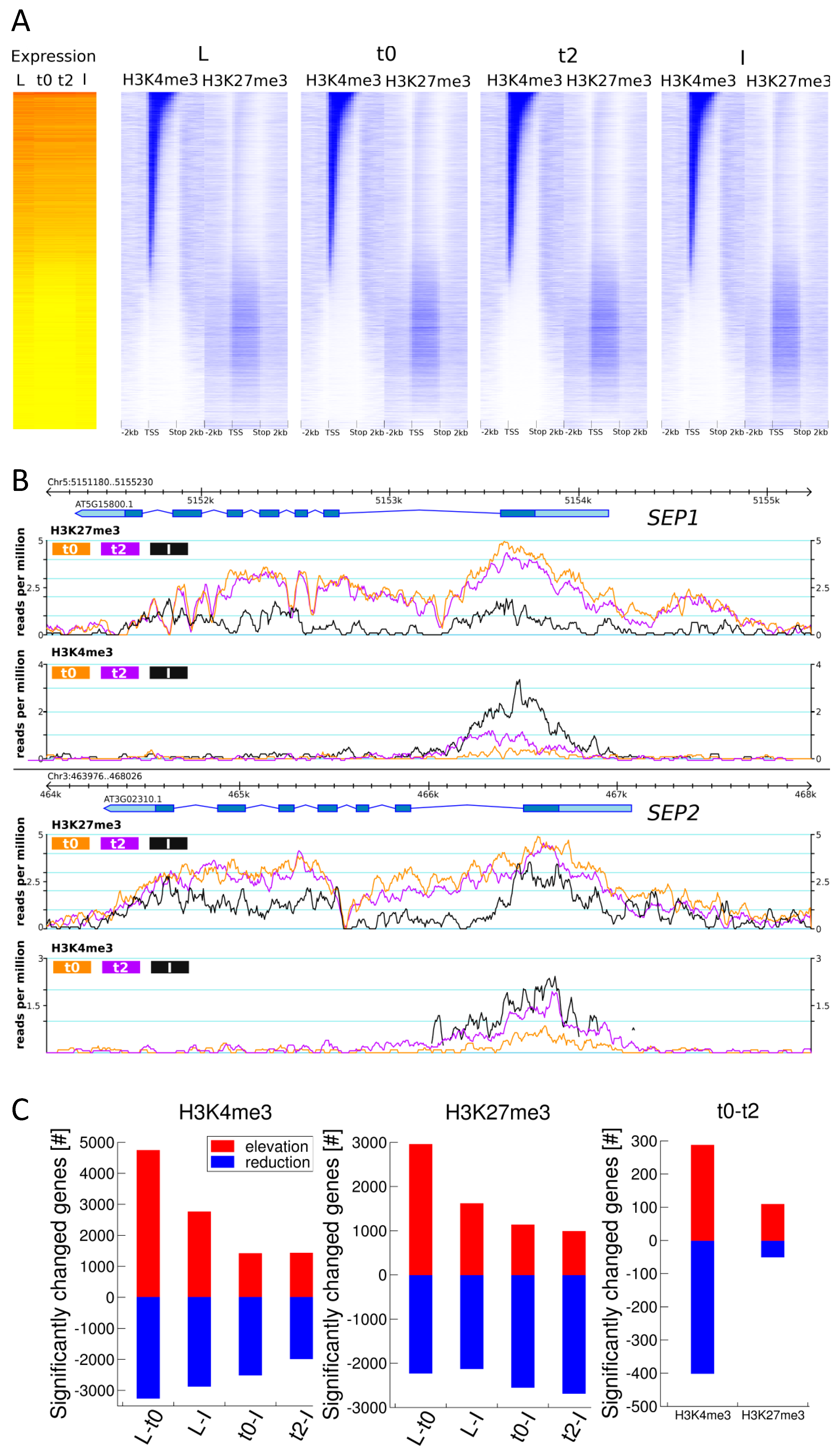
2.2. Meristem and Differentiated Tissues Share Similar Over-All Distribution of H3K27me3 and H3K4me3 Marks
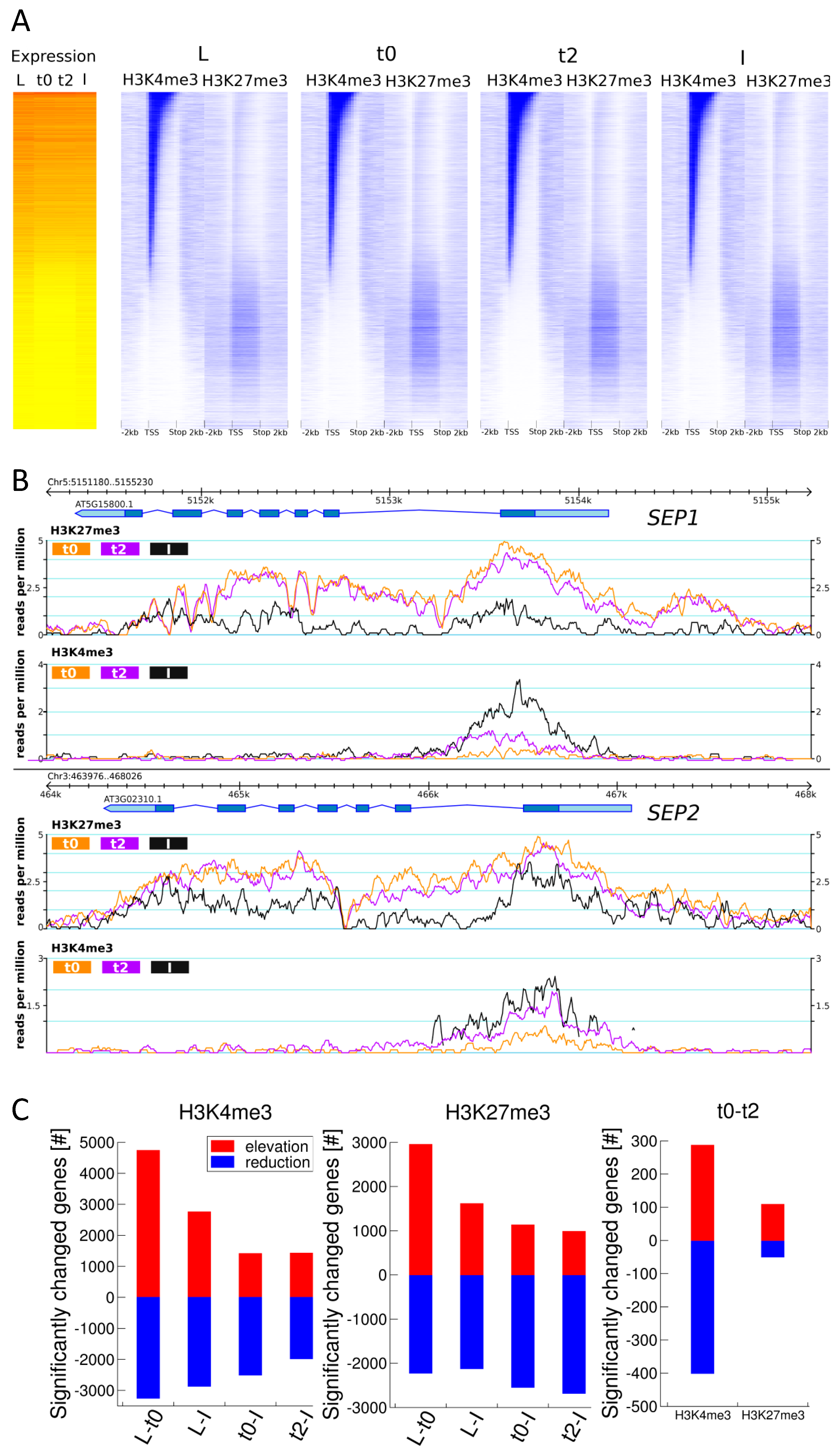
2.3. H3K27me3 and H3K4me3 Levels are Highly Dynamic over the Time Series, in Accordance with Tissue Functionality
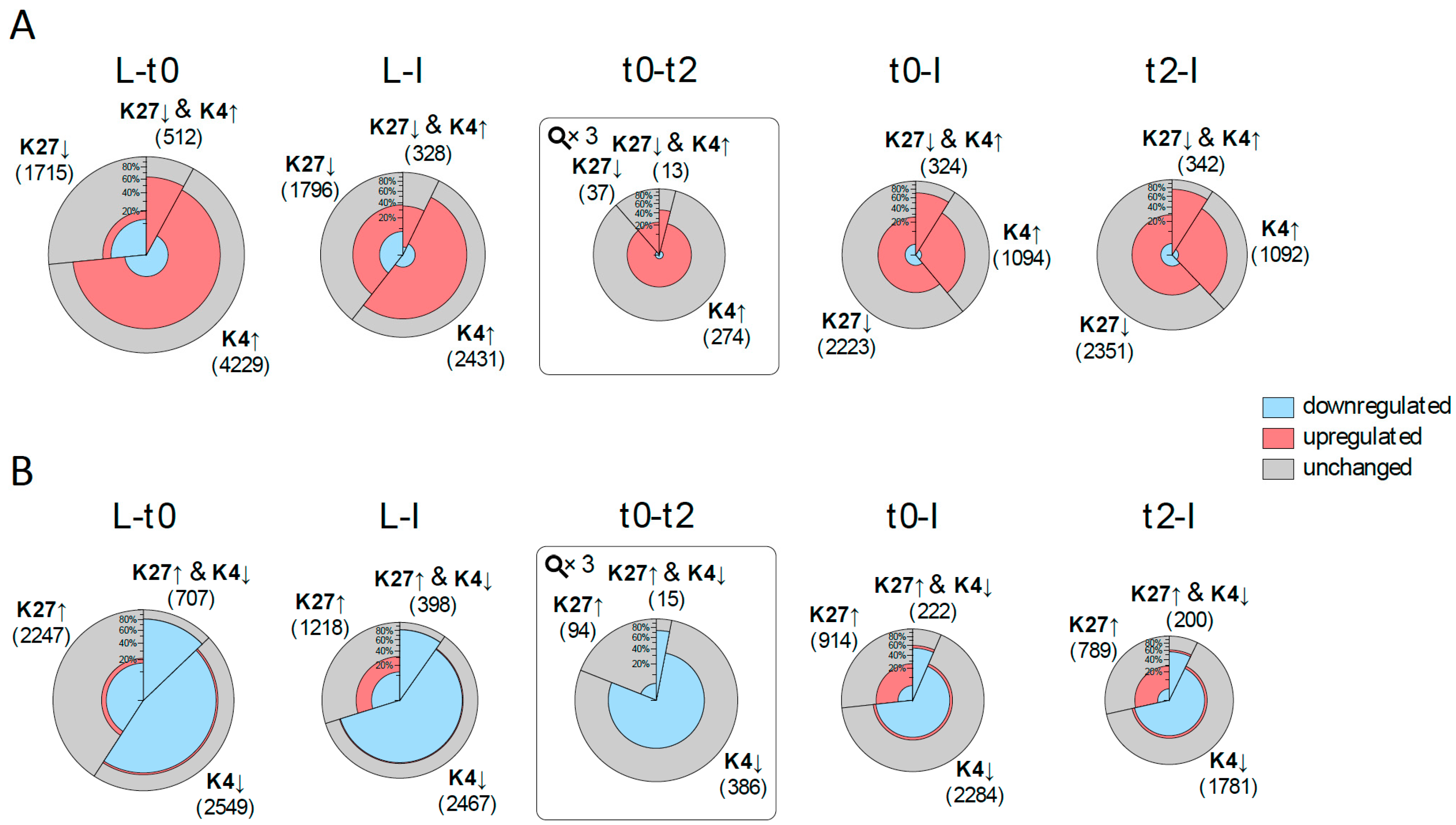
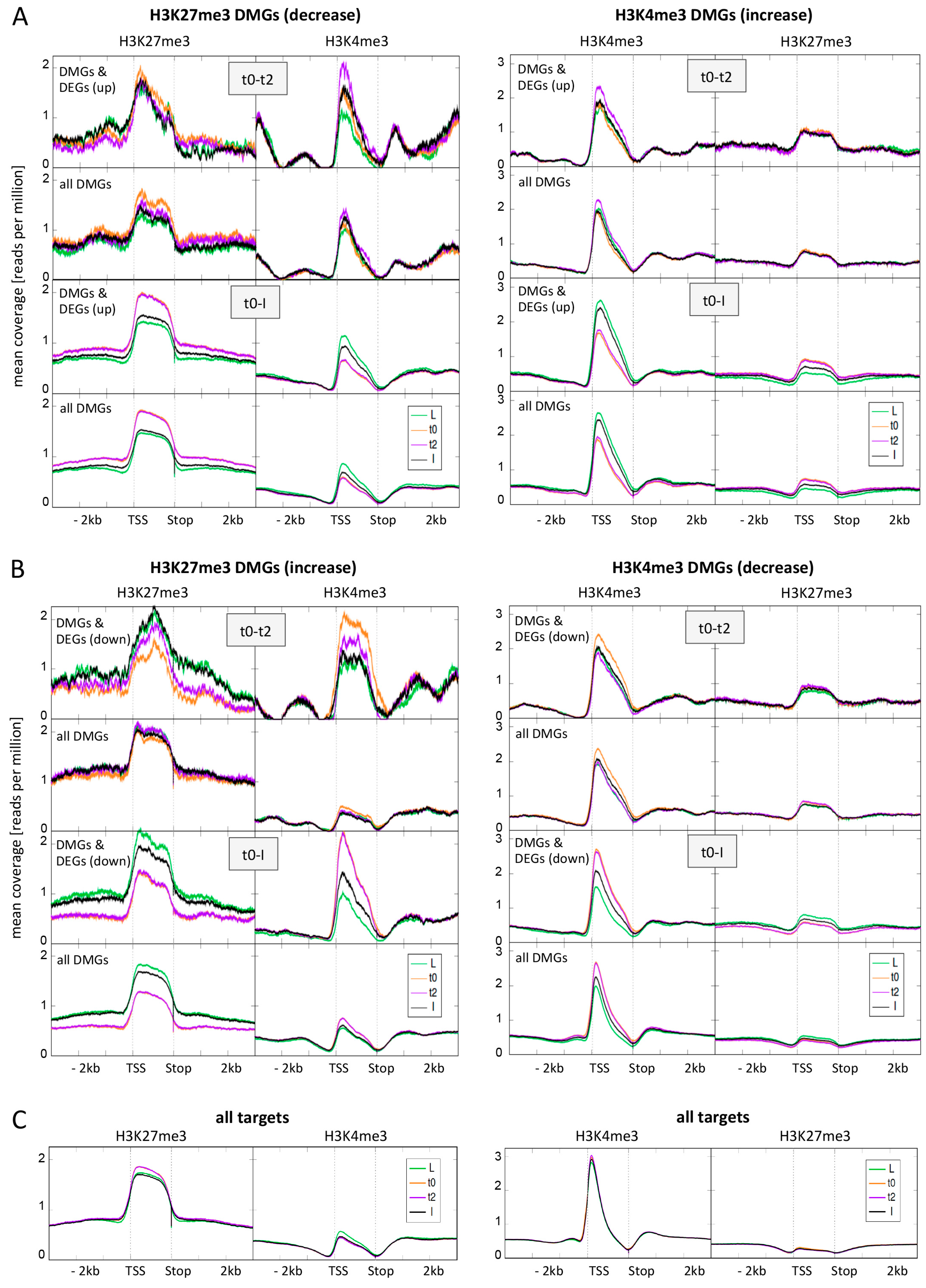
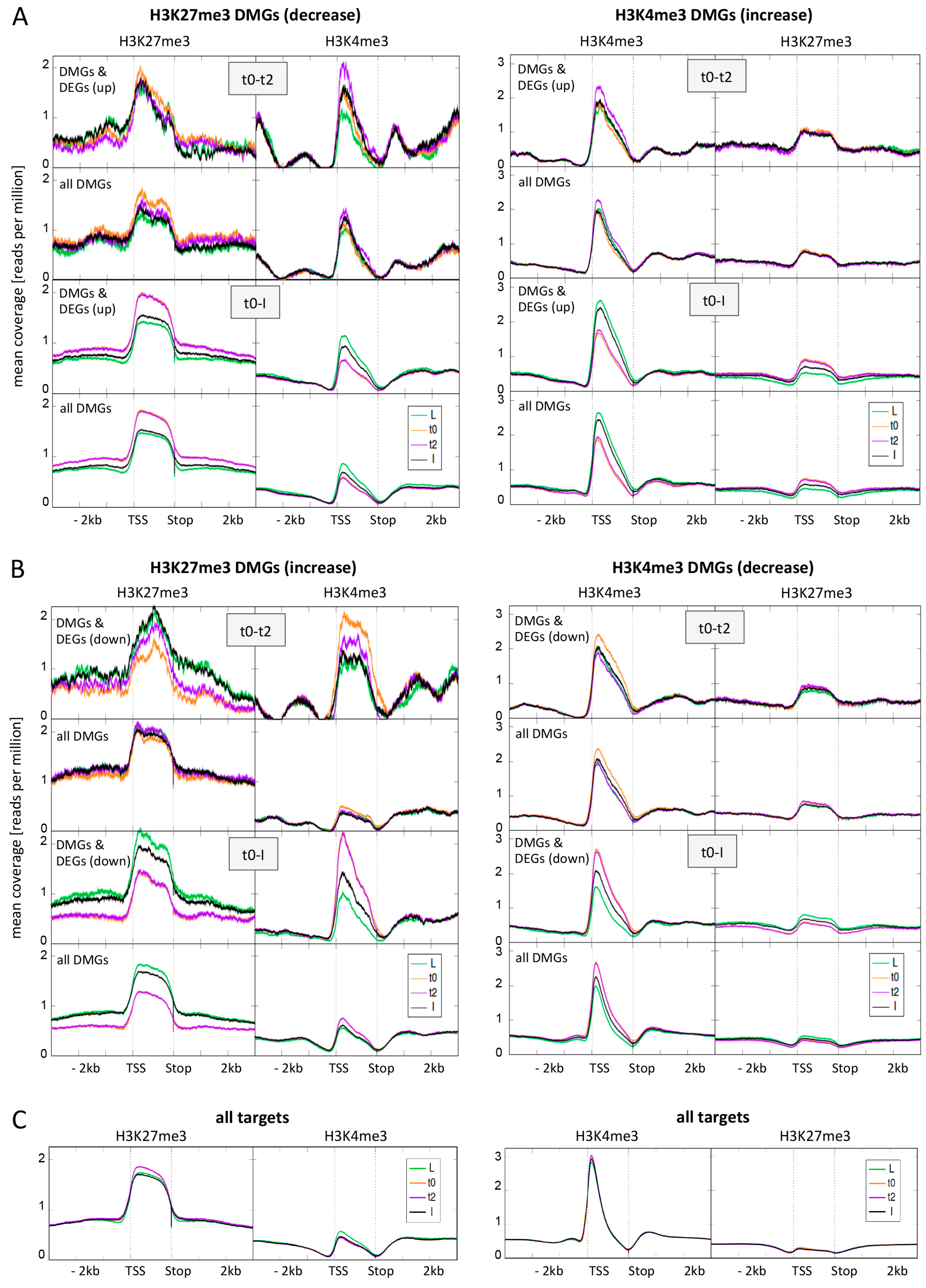
2.4. DMGs Largely Overlap with DEGs and Histone Mark Changes Are Stronger among DEGs
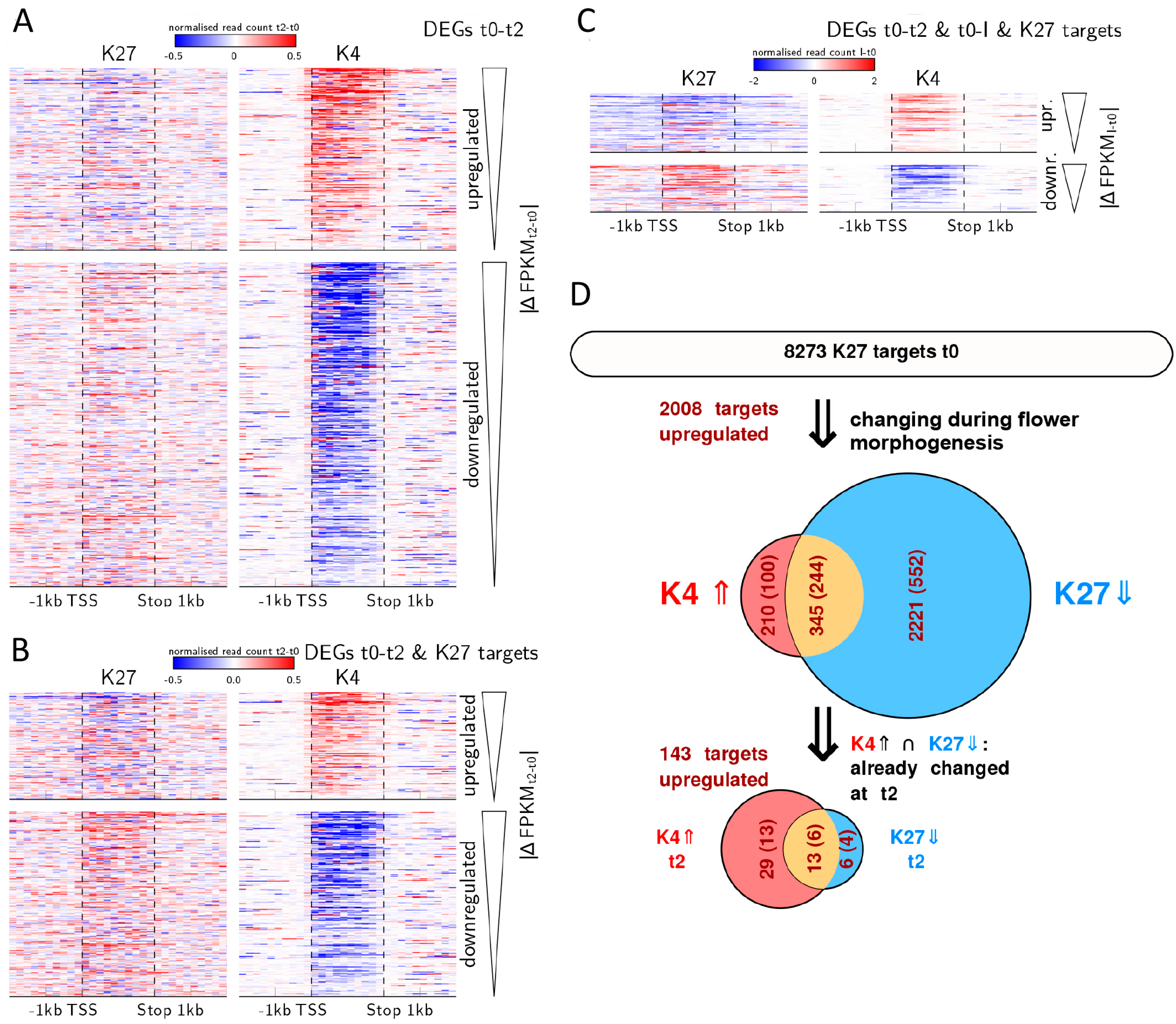
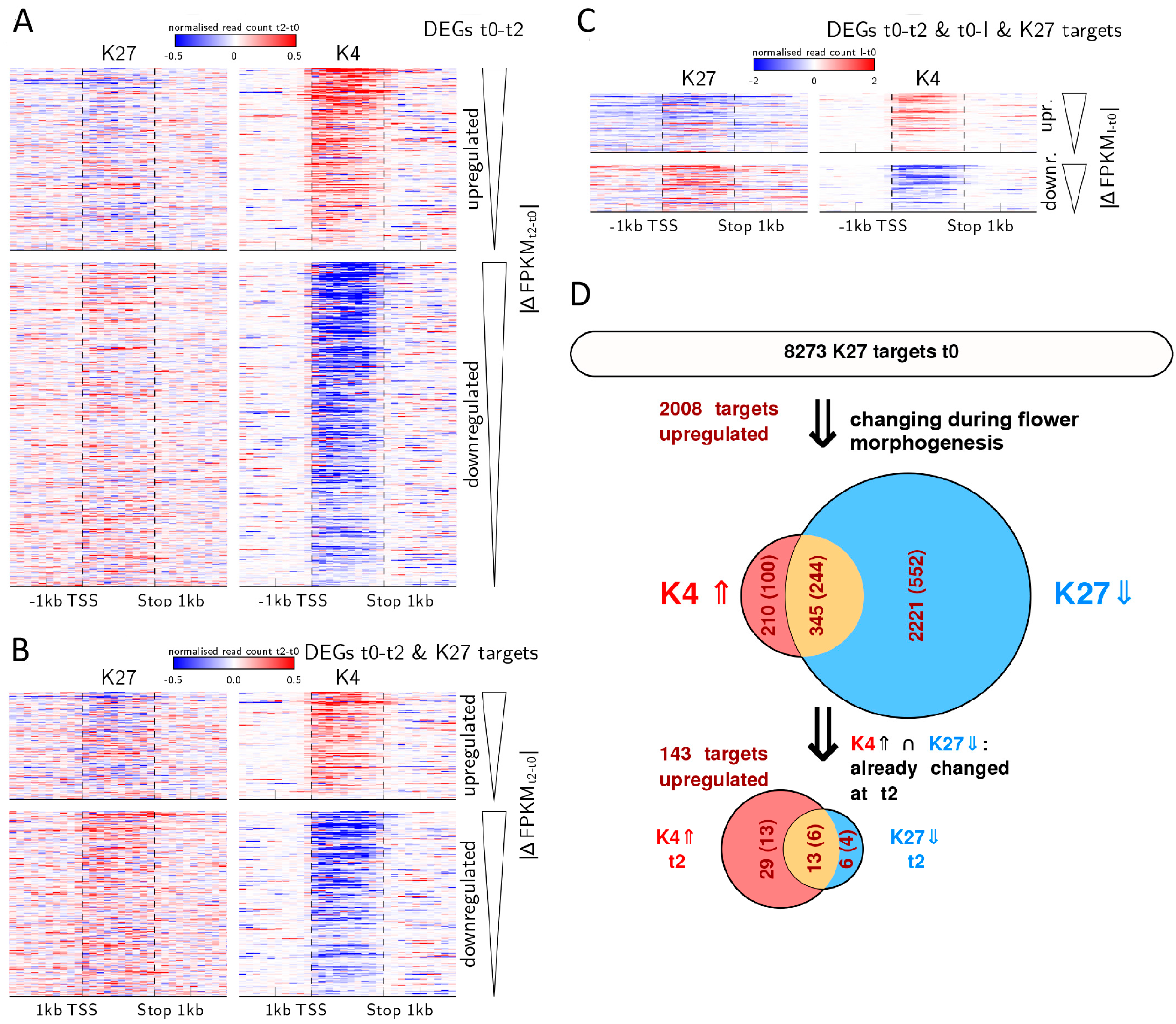
2.5. Expression Changes during Early Flower Morphogenesis Quantitatively Correlate with Changes in H3K4me3 While Changes in H3K27me3 Mostly Occur after Prolonged Expression Changes
2.6. Early Activation of PcG Target Genes is Mainly Accomponied by H3K4me3 Changes
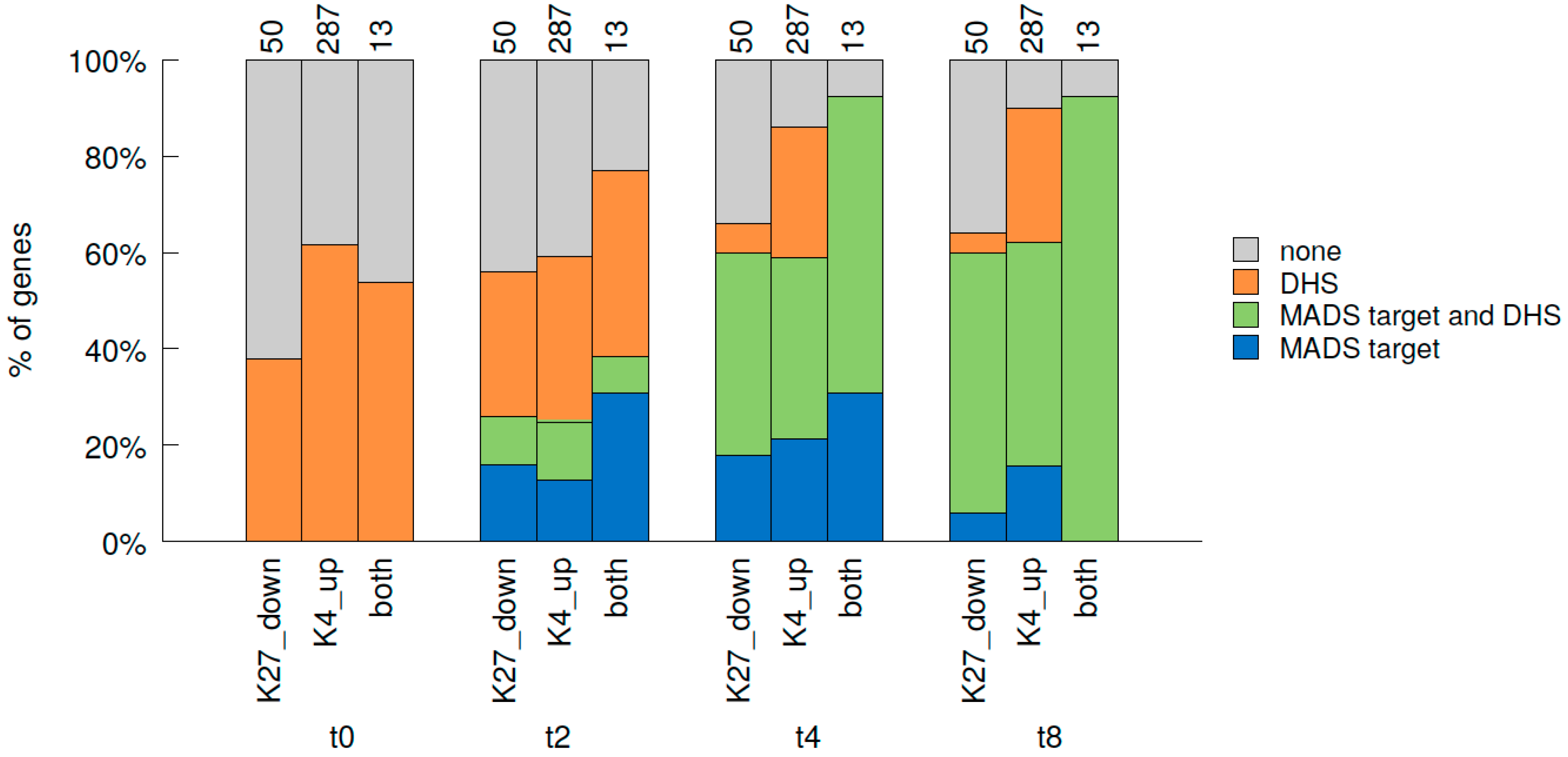
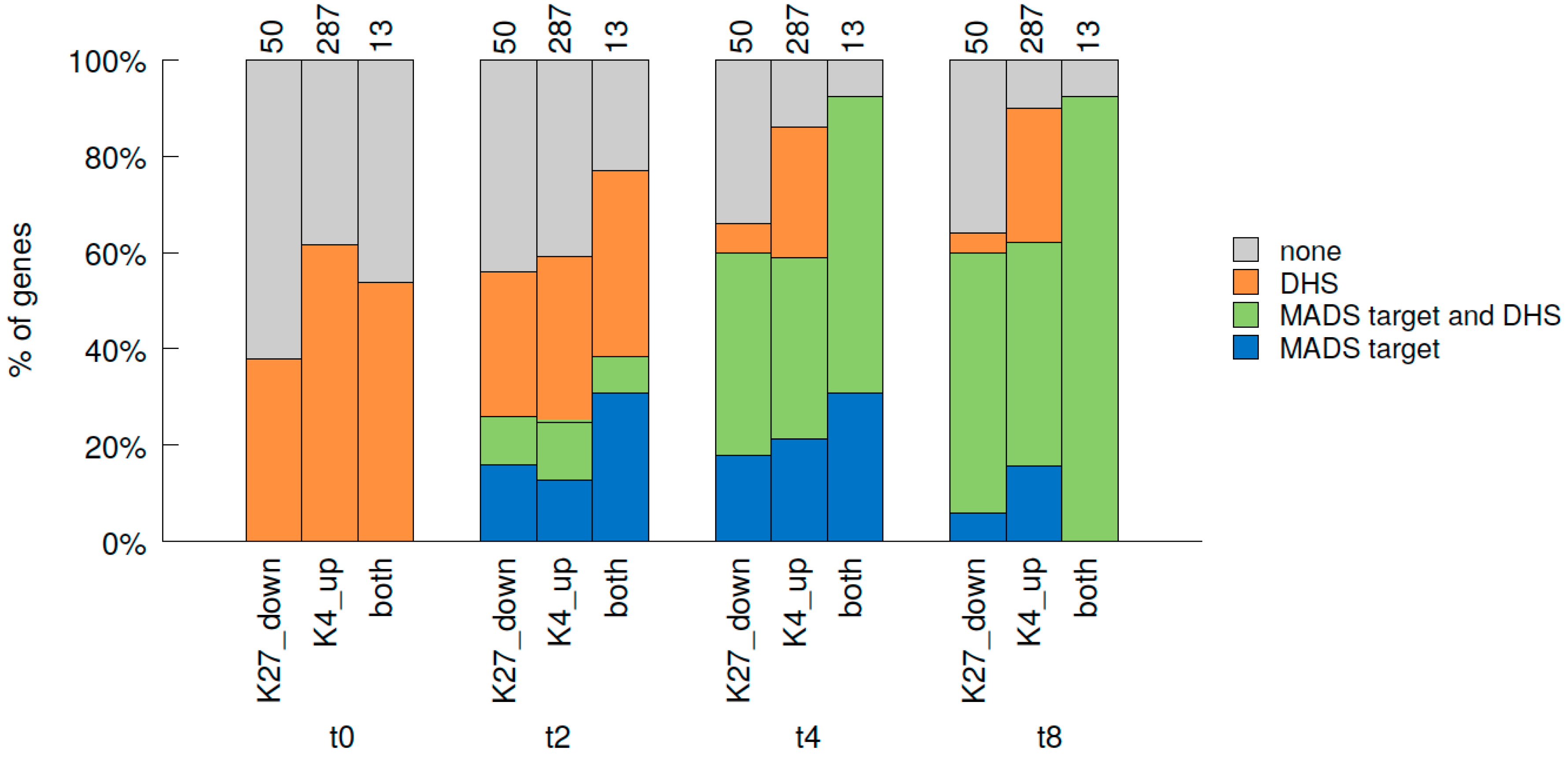
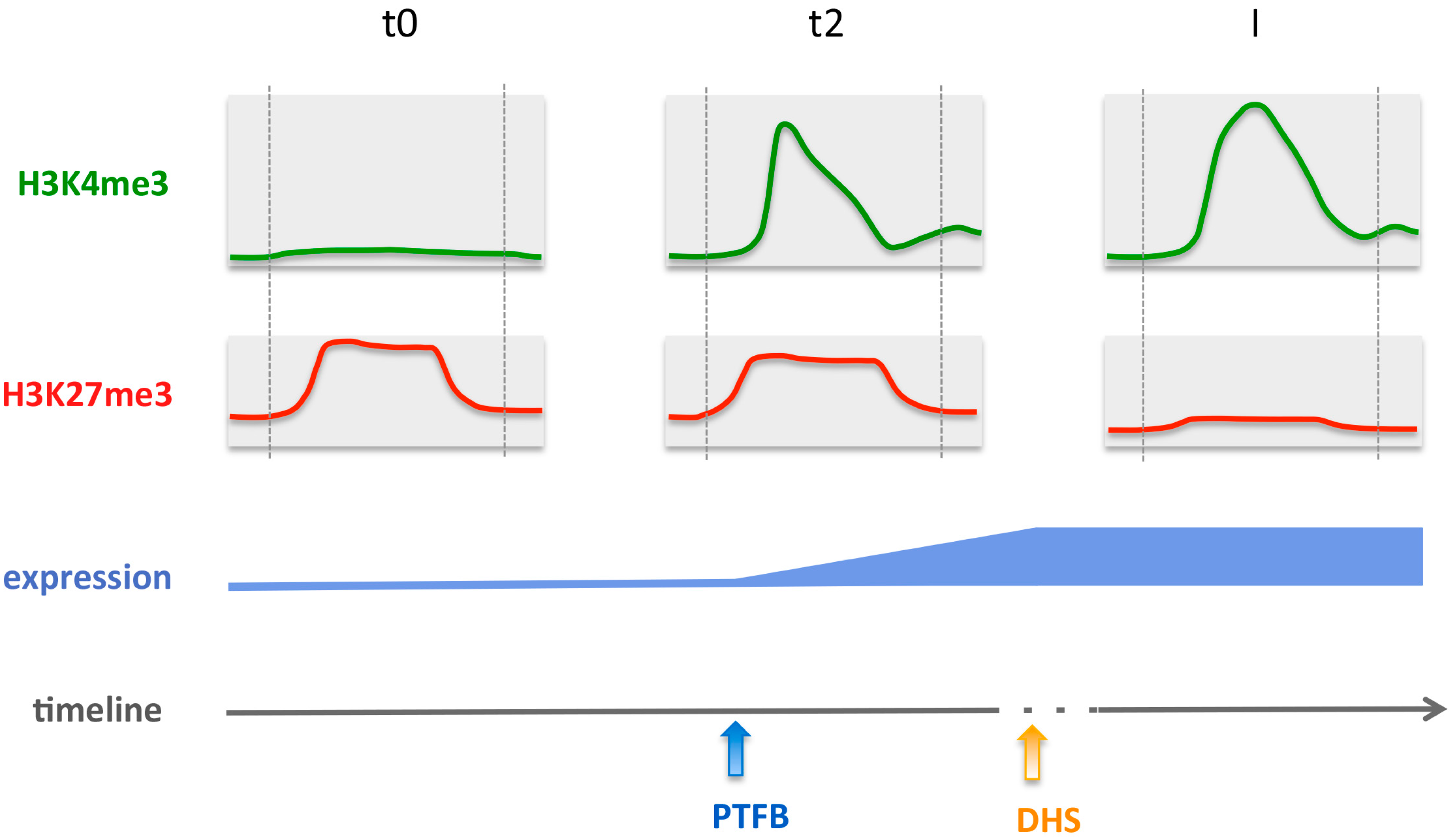
2.7. Relative Dynamics of Histone Marks, MADS TF Binding and Chromatin Opening during Early Stages of Gene Activation
3. Discussion
3.1. Chromatin Marks are Highly Dynamic in Association with Tissue Fate during Flower Morphogenesis
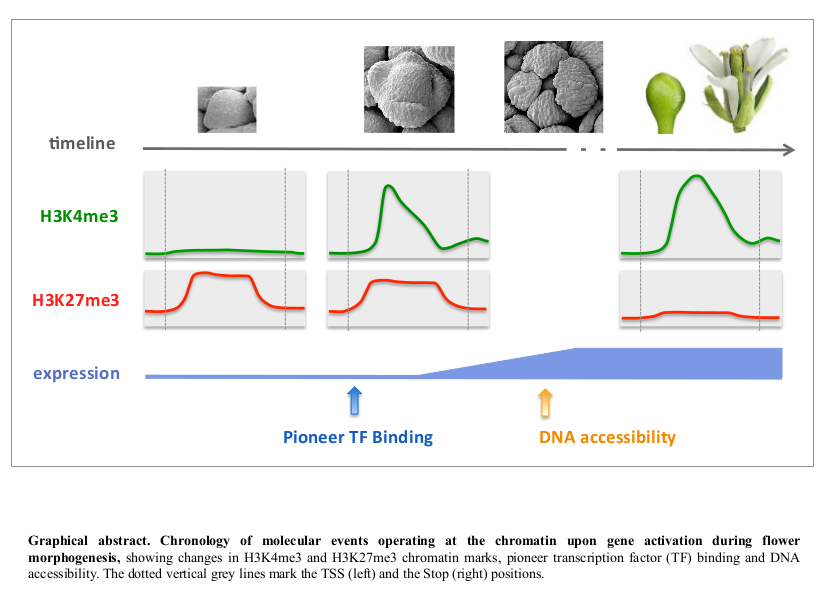
3.2. Integrative Analysis of Chromatin and Expression Datasets Provides Insights on the Mechanistic of Gene Activation, Revealing Unexpected Order of Events
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. Dexamethasone Treatment and Harvesting of Plant Material
4.3. Scanning Electron Microscopy
4.4. Chromatin Immunoprecipitation
4.5. RNA Preparation, Reverse Transcription and Gene Expression Analysis by Semi-qPCR
4.6. Library Preparation and NGS
4.7. ChIP-seq Analysis
4.8. RNA-seq Analysis
4.9. Further Bioinformatics Analyses
4.9.1. Clustering
4.9.2. Gene Ontology Analysis
4.9.3. Visualisation of Data in a Genome Browser
4.9.4. Calculation of average histone mark abundance patterns and heat maps over a locus
4.9.5. Pearson Product-Moment Correlation Coefficient
4.10. Availability of Supporting Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AG | AGAMOUS |
| AGL15 | AGAMOUS-Like 15 |
| AIL6 | AINTEGUMENTA-LIKE 6 |
| AP1-3 | APETALA 1-3 |
| BXL1 | BETA-XYLOSIDASE 1 |
| CAL | CAULIFLOWER |
| ChIP | chromatin-immunoprecipitation |
| CUP-SHAPED COTYLEDON 1 | CUC1 |
| DEGs | differentially expressed genes |
| DHS | DNase I hypersensitive |
| DMGs | differentially marked genes |
| EF1α | Elongation factor 1α |
| FACS | fluorescence-activated cell sorting |
| FLC | FLOWERING LOCUS C |
| FT | FLOWERING LOCUS T |
| GO | Gene Ontology |
| IM | inflorescence meristem |
| LAC10 | LACCASE 10 |
| LBD39 | LOB DOMAIN-CONTAINING PROTEIN 39 |
| LFY | LEAFY |
| LSH | LIGHT SENSITIVE HYPOCOTYLS |
| PcG | Polycomb Group |
| PI | PISTILLATA |
| PRCs | Polycomb repressive complexes |
| PUP4 | PURINE PERMEASE 4 |
| SAM | shoot apical meristem |
| SDG4 | SET-DOMAIN-GROUP4 |
| SEP1-4 | SEPALLATA 1-4 |
| -seq | high-throughput sequencing |
| SHP2 | SHATTERPROOF 2 |
| SOC1 | SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 |
| SPT | SPATULA |
| SVP | SHORT VEGETATIVE PHASE |
| TBP | TATA-box binding protein |
| TF | transcription factor |
| trxG | trithorax Group |
| TSS | transcription start site |
| UFO | UNUSUAL FLORAL ORGANS |
| ULT1 | ULTRAPETALA 1 |
Appendix A. Additional Text
Appendix A.1. Description of Expression Clusters among Floral Regulator Genes
Appendix A.2. K-Means Clustering of DEGs
Appendix A.3. GO Analysis of DEGs
Appendix A.4. Quality Control for ChIP-seq Experiments
Appendix A.5. DMGs during Early Flower Morphogenesis
Appendix A.6. GO Analysis of DMGs
Appendix A.7. Analysis of DMGs That Are Not DEGs
Appendix B.
Appendix B.1. Supplementary Figure Legends
Appendix B.2. Supplementary Table Legends
References and Note
- Alvarez-Buylla, E.R.; Benitez, M.; Corvera-Poire, A.; Chaos Cador, A.; de Folter, S.; Gamboa de Buen, A.; Garay-Arroyo, A.; Garcia-Ponce, B.; Jaimes-Miranda, F.; Perez-Ruiz, R.V.; et al. Flower development. Arabidopsis Book 2010, 8, e0127. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Pajoro, A.; Angenent, G.C. Regulation of transcription in plants: Mechanisms controlling developmental switches. Nat. Rev. Genet. 2010, 11, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Chahtane, I.; Denay, G.; Engelhorn, J.; Monniaux, M.; Moyroud, E.; Moreau, F.; Carles, C.; Tichtinsky, G.; Zubieta, C.; Parcy, F. Flower development: An integrated view. In From Molecules to Living Organisms: An Interplay Between Biology and Physics; Pebay-Peyroula, E., Nury, H., Parcy, F., Ruigrok, R.W.H., Ziegler, C., Cugliandolo, L.F., Eds.; Oxford University Press: Oxford, UK, 2017; in press. [Google Scholar]
- Engelhorn, J.; Blanvillain, R.; Carles, C.C. Gene activation and cell fate control in plants: A chromatin perspective. Cell. Mol. Life Sci. CMLS 2014, 71, 3119–3137. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V. Polycombing the genome: PcG, trxG, and chromatin silencing. Cell 1998, 93, 333–336. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Clarenz, O.; Cokus, S.; Bernatavichute, Y.V.; Pellegrini, M.; Goodrich, J.; Jacobsen, S.E. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007, 5, e129. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Park, S.; van Nocker, S. Genic and global functions for Paf1C in chromatin modification and gene expression in Arabidopsis. PLoS Genet. 2008, 4, e1000077. [Google Scholar] [CrossRef] [PubMed]
- Roudier, F.; Ahmed, I.; Berard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Martinez, A.M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.E.; Tamkun, J.W. Transcriptional regulation by trithorax-group proteins. Cold Spring Harb. Perspect. Biol. 2014, 6, a019349. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Venegas, R. Regulation by polycomb and trithorax group proteins in Arabidopsis. Arabidopsis Book 2010, 8, e0128. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Hennig, L. Regulation of cell identity by plant Polycomb and trithorax group proteins. Curr. Opin. Genet. Dev. 2010, 20, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.; Goodrich, J. Sweet memories: Epigenetic control in flowering. F1000 Biol. Rep. 2011, 3. [Google Scholar] [CrossRef]
- De la Paz Sanchez, M.; Aceves-Garcia, P.; Petrone, E.; Steckenborn, S.; Vega-Leon, R.; Alvarez-Buylla, E.R.; Garay-Arroyo, A.; Garcia-Ponce, B. The impact of Polycomb group (PcG) and Trithorax group (TrxG) epigenetic factors in plant plasticity. New Phytol. 2015, 208, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Sung, Z.R. PcG and trxG in plants—Friends or foes. Trends Genet. TIG 2015, 31, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Hehenberger, E.; Roszak, P.; Hennig, L.; Kohler, C. H3K27me3 profiling of the endosperm implies exclusion of polycomb group protein targeting by DNA methylation. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Lafos, M.; Kroll, P.; Hohenstatt, M.L.; Thorpe, F.L.; Clarenz, O.; Schubert, D. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011, 7, e1002040. [Google Scholar] [CrossRef] [PubMed]
- Brusslan, J.A.; Rus Alvarez-Canterbury, A.M.; Nair, N.U.; Rice, J.C.; Hitchler, M.J.; Pellegrini, M. Genome-wide evaluation of histone methylation changes associated with leaf senescence in Arabidopsis. PLoS ONE 2012, 7, e33151. [Google Scholar] [CrossRef] [PubMed]
- Brusslan, J.A.; Bonora, G.; Rus-Canterbury, A.M.; Tariq, F.; Jaroszewicz, A.; Pellegrini, M. A Genome-Wide Chronological Study of Gene Expression and Two Histone Modifications, H3K4me3 and H3K9ac, during Developmental Leaf Senescence. Plant Physiol. 2015, 168, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Charron, J.B.; He, H.; Elling, A.A.; Deng, X.W. Dynamic landscapes of four histone modifications during deetiolation in Arabidopsis. Plant Cell 2009, 21, 3732–3748. [Google Scholar] [CrossRef] [PubMed]
- He., C.; Chen, X.; Huang, H.; Xu, L. Reprogramming of H3K27me3 is critical for acquisition of pluripotency from cultured Arabidopsis tissues. PLoS Genet. 2012, 8, e1002911. [Google Scholar] [CrossRef] [PubMed]
- Adrian, J.; Farrona, S.; Reimer, J.J.; Albani, M.C.; Coupland, G.; Turck, F. cis-Regulatory elements and chromatin state coordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell 2010, 22, 1425–1440. [Google Scholar] [CrossRef] [PubMed]
- Buzas, D.M.; Robertson, M.; Finnegan, E.J.; Helliwell, C.A. Transcription-dependence of histone H3 lysine 27 trimethylation at the Arabidopsis polycomb target gene FLC. Plant J. Cell Mol. Biol. 2011, 65, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Wellmer, F.; Alves-Ferreira, M.; Dubois, A.; Riechmann, J.L.; Meyerowitz, E.M. Genome-wide analysis of gene expression during early Arabidopsis flower development. PLoS Genet. 2006, 2, e117. [Google Scholar] [CrossRef]
- Kaufmann, K.; Wellmer, F.; Muino, J.M.; Ferrier, T.; Wuest, S.E.; Kumar, V.; Serrano-Mislata, A.; Madueno, F.; Krajewski, P.; Meyerowitz, E.M.; et al. Orchestration of floral initiation by APETALA1. Science 2010, 328, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Smaczniak, C.; Immink, R.G.; Muino, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.D.; Liu, S.; Westphal, A.H.; Boeren, S.; et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2012, 109, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Pajoro, A.; Madrigal, P.; Muino, J.M.; Matus, J.T.; Jin, J.; Mecchia, M.A.; Debernardi, J.M.; Palatnik, J.F.; Balazadeh, S.; Arif, M.; et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 2014, 15, R41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.S.; Huang, J.; Ito, T. Functional roles of histone modification, chromatin remodeling and microRNAs in Arabidopsis flower development. Int. Rev. Cell Mol. Biol. 2013, 305, 115–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Fromm, M.; Avramova, Z. H3K27me3 and H3K4me3 chromatin environment at super-induced dehydration stress memory genes of Arabidopsis thaliana. Mol. Plant 2014, 7, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Burkle, L.; Andre, B.; Kuhn, C.; Rentsch, D.; Brandl, B.; Frommer, W.B. A new family of high-affinity transporters for adenine, cytosine, and purine derivatives in Arabidopsis. Plant Cell 2000, 12, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Madrid, R.; Mestres, D.; Marinho, P.; Jacquot, J.P.; Decottignies, P.; Miginiac-Maslow, M.; Meyer, Y. Evidence for five divergent thioredoxin h sequences in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1995, 92, 5620–5624. [Google Scholar] [CrossRef] [PubMed]
- Laloi, C.; Mestres-Ortega, D.; Marco, Y.; Meyer, Y.; Reichheld, J.P. The Arabidopsis cytosolic thioredoxin h5 gene induction by oxidative stress and its W-box-mediated response to pathogen elicitor. Plant Physiol. 2004, 134, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Sarry, J.E.; Kuhn, L.; Ducruix, C.; Lafaye, A.; Junot, C.; Hugouvieux, V.; Jourdain, A.; Bastien, O.; Fievet, J.B.; Vailhen, D.; et al. The early responses of Arabidopsis thaliana cells to cadmium exposure explored by protein and metabolite profiling analyses. Proteomics 2006, 6, 2180–2198. [Google Scholar] [CrossRef] [PubMed]
- Sweat, T.A.; Wolpert, T.J. Thioredoxin h5 is required for victorin sensitivity mediated by a CC-NBS-LRR gene in Arabidopsis. Plant Cell 2007, 19, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mena, C.; de Folter, S.; Costa, M.M.; Angenent, G.C.; Sablowski, R. Transcriptional program controlled by the floral homeotic gene AGAMOUS during early organogenesis. Development 2005, 132, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Liu, Q.; Dubouzet, J.G.; Abe, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 2002, 290, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Dinant, S.; Clark, A.M.; Zhu, Y.; Vilaine, F.; Palauqui, J.C.; Kusiak, C.; Thompson, G.A. Diversity of the superfamily of phloem lectins (phloem protein 2) in angiosperms. Plant Physiol. 2003, 131, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yanofsky, M.F.; Meyerowitz, E.M. AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 1991, 5, 484–495. [Google Scholar]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, M.R.; Bedgar, D.L.; Moinuddin, S.G.; Cardenas, C.L.; Davin, L.B.; Kang, C.; Lewis, N.G. Functional reclassification of the putative cinnamyl alcohol dehydrogenase multigene family in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Smyth, D.R. CRABS CLAW and SPATULA, two Arabidopsis genes that control carpel development in parallel with AGAMOUS. Development 1999, 126, 2377–2386. [Google Scholar] [PubMed]
- Heisler, M.G.; Atkinson, A.; Bylstra, Y.H.; Walsh, R.; Smyth, D.R. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development 2001, 128, 1089–1098. [Google Scholar] [PubMed]
- Turlapati, P.V.; Kim, K.W.; Davin, L.B.; Lewis, N.G. The laccase multigene family in Arabidopsis thaliana: Towards addressing the mystery of their gene function(s). Planta 2011, 233, 439–470. [Google Scholar] [CrossRef] [PubMed]
- Krizek, B.A. Aintegumenta and Aintegumenta-Like6 regulate auxin-mediated flower development in Arabidopsis. BMC Res. Notes 2011, 4, 176. [Google Scholar] [CrossRef] [PubMed]
- Krizek, B.A.; Eaddy, M. AINTEGUMENTA-LIKE6 regulates cellular differentiation in flowers. Plant Mol. Biol. 2012, 78, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.; Alvarez, J.; Smyth, D.R.; Yanofsky, M.F.; Meyerowitz, E.M. LFY controls floral meristem identity in Arabidopsis. Cell 1992, 69, 843–859. [Google Scholar] [CrossRef]
- Blazquez, M.A.; Soowal, L.N.; Lee, I.; Weigel, D. LEAFY expression and flower initiation in Arabidopsis. Development 1997, 124, 3835–3844. [Google Scholar] [PubMed]
- Parcy, F.; Nilsson, O.; Busch, M.A.; Lee, I.; Weigel, D. A genetic framework for floral patterning. Nature 1998, 395, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.X.; Wang, Y.X.; Liu, X.F.; Li, J.Y. Arabidopsis RAV1 is down-regulated by brassinosteroid and may act as a negative regulator during plant development. Cell Res. 2004, 14, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.G.; Cook, D.; Thomashow, M.F. Low temperature induction of Arabidopsis CBF1, 2, and 3 is gated by the circadian clock. Plant Physiol. 2005, 137, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Petricka, J.J.; Clay, N.K.; Nelson, T.M. Vein patterning screens and the defectively organized tributaries mutants in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2008, 56, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Hanano, K.; Kariya, A.; Shimizu, S.; Zhao, L.; Matsui, M.; Tasaka, M.; Aida, M. CUP-SHAPED COTYLEDON1 transcription factor activates the expression of LSH4 and LSH3, two members of the ALOG gene family, in shoot organ boundary cells. Plant J. Cell Mol. Biol. 2011, 66, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Zambryski, P.C. Organ boundary1 defines a gene expressed at the junction between the shoot apical meristem and lateral organs. Proc. Natl. Acad. Sci. USA 2011, 108, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Nakazawa, M.; Takase, T.; Manabe, K.; Kobayashi, M.; Seki, M.; Shinozaki, K.; Matsui, M. Overexpression of LSH1, a member of an uncharacterised gene family, causes enhanced light regulation of seedling development. Plant J. Cell Mol. Biol. 2004, 37, 694–706. [Google Scholar] [CrossRef]
- Joshi, V.; Laubengayer, K.M.; Schauer, N.; Fernie, A.R.; Jander, G. Two Arabidopsis threonine aldolases are nonredundant and compete with threonine deaminase for a common substrate pool. Plant Cell 2006, 18, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakazawa, M.; Kawashima, M.; Iizumi, H.; Kuroda, H.; Kondou, Y.; Tsuhara, Y.; Suzuki, K.; Ishikawa, A.; Seki, M.; et al. The FOX hunting system: An alternative gain-of-function gene hunting technique. Plant J. Cell Mol. Biol. 2006, 48, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Flematti, G.R.; Riseborough, J.A.; Ghisalberti, E.L.; Dixon, K.W.; Smith, S.M. Karrikins enhance light responses during germination and seedling development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 7095–7100. [Google Scholar] [CrossRef] [PubMed]
- Cartagena, J.A.; Matsunaga, S.; Seki, M.; Kurihara, D.; Yokoyama, M.; Shinozaki, K.; Fujimoto, S.; Azumi, Y.; Uchiyama, S.; Fukui, K. The Arabidopsis SDG4 contributes to the regulation of pollen tube growth by methylation of histone H3 lysines 4 and 36 in mature pollen. Dev. Biol. 2008, 315, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.T.; Moylan, E.C.; Langdale, J.A. GLK transcription factors regulate chloroplast development in a cell-autonomous manner. Plant J. Cell Mol. Biol. 2008, 56, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Arsovski, A.A.; Popma, T.M.; Haughn, G.W.; Carpita, N.C.; McCann, M.C.; Western, T.L. AtBXL1 encodes a bifunctional beta-D-xylosidase/alpha-L-arabinofuranosidase required for pectic arabinan modification in Arabidopsis mucilage secretory cells. Plant Physiol. 2009, 150, 1219–1234. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Primavesi, L.; Bishopp, A.; Roberts, G.; Doonan, J.; Jenuwein, T.; Goodrich, J. Silencing by plant Polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J. 2006, 25, 4638–4649. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Deng, S.; Wang, H.; Ye, J.; Wu, H.W.; Sun, H.X.; Chua, N.H. CURLY LEAF Regulates Gene Sets Coordinating Seed Size and Lipid Biosynthesis. Plant Physiol. 2016, 171, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.R.; Bowman, J.L.; Meyerowitz, E.M. Early flower development in Arabidopsis. Plant Cell 1990, 2, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.; Das, P.; Mirabet, V.; Moscardi, E.; Traas, J.; Verdeil, J.L.; Malandain, G.; Godin, C. Imaging plant growth in 4D: Robust tissue reconstruction and lineaging at cell resolution. Nat. Methods 2010, 7, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Looi, L.S.; Guo, S.; He, Z.; Gan, E.S.; Huang, J.; Xu, Y.; Wee, W.Y.; Ito, T. Timing mechanism dependent on cell division is invoked by Polycomb eviction in plant stem cells. Science 2014, 343, 1248559. [Google Scholar] [CrossRef] [PubMed]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stutzer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 2011, 42, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ndamukong, I.; Xu, Z.; Lapko, H.; Fromm, M.; Avramova, Z. ATX1-generated H3K4me3 is required for efficient elongation of transcription, not initiation, at ATX1-regulated genes. PLoS Genet. 2012, 8, e1003111. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Sidote, D.J.; Zhang, Y.; Kerstetter, R.A.; Michael, T.P.; Lam, E. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J. Cell Mol. Biol. 2013, 73, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Sequeira-Mendes, J.; Araguez, I.; Peiro, R.; Mendez-Giraldez, R.; Zhang, X.; Jacobsen, S.E.; Bastolla, U.; Gutierrez, C. The Functional Topography of the Arabidopsis Genome Is Organized in a Reduced Number of Linear Motifs of Chromatin States. Plant Cell 2014, 26, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Alvarez, J.; Weigel, D.; Meyerowitz, E. M.; Smyth, D. R. Control of flower development in Arabidopsis thaliana by APETALA 1 and interacting genes. Development 1993, 119, 721–743. [Google Scholar]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genes directing flower development in Arabidopsis. Plant Cell 1989, 1, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Carles, C.C.; Fletcher, J.C. The SAND domain protein ULTRAPETALA1 acts as a trithorax group factor to regulate cell fate in plants. Genes Dev. 2009, 23, 2723–2728. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Muino, J.M.; Osteras, M.; Farinelli, L.; Krajewski, P.; Angenent, G.C. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 2010, 5, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.; Mathieu, J.; Dinh, T.T.; Ott, F.; Lanz, C.; Wollmann, H.; Chen, X.; Schmid, M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 2010, 22, 2156–2170. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.G.; Pose, D.; Ferrario, S.; Ott, F.; Kaufmann, K.; Valentim, F.L.; de Folter, S.; van der Wal, F.; van Dijk, A.D.; Schmid, M.; et al. Characterization of SOC1's central role in flowering by the identification of its upstream and downstream regulators. Plant Physiol. 2012, 160, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Picard-tools dfpsnin, website now moved to http://broadinstitute.github.io/picard, version used is 1.93.
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Engelhorn, J.; Turck, F. Metaanalysis of ChIP-chip data. Methods Mol. Biol. 2010, 631, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Provart, N.; Zhu, T. A browser-based functional classification SuperViewer for Arabidopsis genomics. Curr. Comput. Mol. Biol. 2003, 2003, 271–272. [Google Scholar]
- Al-Shahrour, F.; Minguez, P.; Tarraga, J.; Medina, I.; Alloza, E.; Montaner, D.; Dopazo, J. FatiGO +: A functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res. 2007, 35, W91–W96. [Google Scholar] [CrossRef] [PubMed]
- Medina, I.; Carbonell, J.; Pulido, L.; Madeira, S.C.; Goetz, S.; Conesa, A.; Tarraga, J.; Pascual-Montano, A.; Nogales-Cadenas, R.; Santoyo, J.; et al. Babelomics: An integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010, 38, W210–W213. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Zweig, A.S.; Barber, G.; Hinrichs, A.S.; Karolchik, D. BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 2010, 26, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.D. Using GBrowse 2.0 to visualize and share next-generation sequence data. Brief Bioinform. 2013, 14, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Farrona, S.; Thorpe, F.L.; Engelhorn, J.; Adrian, J.; Dong, X.; Sarid-Krebs, L.; Goodrich, J.; Turck, F. Tissue-specific expression of FLOWERING LOCUS T in Arabidopsis is maintained independently of polycomb group protein repression. Plant Cell 2011, 23, 3204–3214. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activation | ||||||
| L < I | L < t0 | L < t2 | t0 < I | t2 < I | t0 < t2 | |
| L < I | 4910 | |||||
| L < t0 | 2835 | 4215 | ||||
| L < t2 | 2832 | 3865 | 4246 | |||
| t0 < I | 1496 | 154 | 181 | 3915 | ||
| t2 < I | 1581 | 188 | 144 | 3252 | 4530 | |
| t0 < t2 | 96 | 61 | 118 | 131 | 59 | 251 |
| Repression | ||||||
| L > I | L > t0 | L > t2 | t0 > I | t2 > I | t0 > t2 | |
| L > I | 4629 | |||||
| L > t0 | 3913 | 5683 | ||||
| L > t2 | 3966 | 5170 | 5964 | |||
| t0 > I | 186 | 65 | 98 | 1252 | ||
| t2 > I | 145 | 54 | 40 | 930 | 1365 | |
| t0 > t2 | 194 | 199 | 281 | 105 | 33 | 448 |
| Activation | ||||
| Identifier | Name | Domains/Family | Function | References |
| At1g30840 | PURINE PERMEASE 4 (PUP4) | Member of a family of proteins related to PUP1, a purine transporter | putative purine transporter | [33] |
| At1g45145 | LOCUS OF INSENSITIVITY TO VICTORIN 1 (LIV1), THIOREDOXIN H-TYPE 5 (TRX-H5) | cytosolic thioredoxin | response to biotic and abiotic stimuli | [34,35,36,37] |
| At1g47610 | Transducin/WD40 repeat-like superfamily protein | unknown, known to be upregulated upon induction of AG function after 3 days | [38] | |
| At1g75490 | DEHYDRATION-RESPONSIVE ELEMENT BINDING (DREB) A-2 ERF/AP2 TF family | DREB2A and DREB2B involved in drought response | [39,40] | |
| At1g80110 | PHLOEM PROTEIN 2-B11 (PP2-B11) | putative carbohydrate binding | [41] | |
| At3g02310 | SEPALLATA 2 (SEP2) | MADS TF | floral organ identity specification | [42,43] |
| At4g32990 | Transducin/WD40 repeat-like superfamily | |||
| At4g34230 | CINNAMYL ALCOHOL DEHYDROGENASE 5 (CAD5) | catalytically active cinnamyl alcohol dehydrogenase | [44] | |
| At4g36930 | SPATULA (SPT) | bHLH TF | carpel development and seed dormancy regulation | [45,46] |
| LACCASE 10 (LAC10) | putative laccase | [47] | ||
| At5g10510 | AINTEGUMENTA-LIKE 6 (AIL6), PLETHORA 3 (PLT3) | AP2 domain TF | proliferation and differentiation in flowers, root stem cell identity, promote PIN1 expression and modulate local auxin production in phyllotaxis of the apex | [48,49] |
| At5g15800 | SEPALLATA 1 (SEP1) | MADS TF | floral organ identity specification | [42,43] |
| At5g61850 | LEAFY (LFY) | floral meristem identity control | [50,51,52] | |
| Repression | ||||
| Identifier | Name | Domains/Family | Function | References |
| At1g05370 | Sec14p-like | phosphatidylinositol transfer | ||
| At1g13260 | ETHYLENE RESPONSE DNA BINDING FACTOR 4 (EDF4), RELATED TO ABI3/VP11 (RAV1) | AP2/B3 domain TF | upregulated in low temperatures, has circadian regulation and may function as a negative growth regulator | [53,54] |
| At1g13290 | DEFECTIVELY ORGANIZED TRIBUTARIES 5 (DOT5) WIP DOMAIN PROTEIN 6 (WIP6) | C2H2-ZF (type IIIA, subclass A1d) with a WIP domain. | venation in leaves, petiol development and phyllotaxy regulation | [55] |
| At2g28550 | RELATED TO AP2.7 (RAP2.7), TARGET OF EARLY ACTIVATION TAGGED (EAT) 1 (TOE1) | AP2 TF | negative regulation of floral transition | [56] |
| At2g31160 | LIGHT SENSITIVE HYPOCOTYLS 3 (LSH3), ORGAN BOUNDARY 1 (OBO1) | LSH1 family protein | may suppress organ differentiation in the boundary region of the SAM | [57,58,59] |
| At2g34510 | unknown | expressed | ||
| At3g04510 | LIGHT SENSITIVE HYPOCOTYLS 2 (LSH2) | LSH1 family | LHS1 regulates light regulation of seedling development | [57,58,59] |
| At3g04520 | THREONINE ALDOLASE 2 (THA2) | threonine aldolase | threonine degradation to glycine | [60] |
| At3g55240 | overexpression leads to Pseudo-Etiolation in Light | [61] | ||
| At4g24110 | unknown | expressed, responds to karrikins | [62] | |
| At4g37540 | LOB DOMAIN-CONTAINING PROTEIN 39 (LBD39) | LOB | downregulated in SET DOMAIN GROUP 4 mutant flowers (SDG4 maintain methylated histone H3 K4 and K36 levels) | [63] |
| At5g06380 | expressed protein | unknown | ||
| At5g28490 | LIGHT-DEPENDENT SHORT HYPOCOTYLS 1 (LSH1), ORGAN BOUNDARY 2 (OBO2) | LSH1 family | light regulation of seedling development in a phytochrome-dependent manner | [57,58,59] |
| At5g44190 | GBF’S PRO-RICH REGION-INTERACTING FACTOR 2 (GPRI2), GOLDEN2-LIKE 2 (GLK2) | GOLDEN2-like TF | regulation of chloroplast development in a cell-autonomous manner, regulation of the expression of the photosynthetic apparatus, negative regulation of floral transition | [64] |
| At5g49360 | BETA-XYLOSIDASE 1 (BXL1) | glycosyl hydrolase family 3 | β-D-xylosidase/α-L-arabinofuranosidase of the extracellular matrix required for pectic arabinan modification. expressed in tissues with secondary wall thickening, involved in seed germination | [65] |
| PCC | Δ Normalised Read Count | ||||
|---|---|---|---|---|---|
| H3K27me3 | H3K4me3 | H3K27me3 | H3K4me3 | ||
| up-regulated DEGs | t0-t2 | −0.104 | 0.456 | −0.004 | 0.139 |
| t0-t2, K27 targets | −0.152 | 0.420 | −0.002 | 0.110 | |
| t0-t2 & t0-I, K27 targets | −0.248 | 0.360 | −0.230 | 0.289 | |
| down-regulated DEGs | t0-t2 | −0.012 | 0.420 | 0.032 | −0.190 |
| t0-t2, K27 targets | −0.197 | 0.389 | 0.062 | −0.169 | |
| t0-t2 & t0-I, K27 targets | −0.284 | 0.467 | 0.428 | −0.448 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engelhorn, J.; Blanvillain, R.; Kröner, C.; Parrinello, H.; Rohmer, M.; Posé, D.; Ott, F.; Schmid, M.; Carles, C.C. Dynamics of H3K4me3 Chromatin Marks Prevails over H3K27me3 for Gene Regulation during Flower Morphogenesis in Arabidopsis thaliana. Epigenomes 2017, 1, 8. https://doi.org/10.3390/epigenomes1020008
Engelhorn J, Blanvillain R, Kröner C, Parrinello H, Rohmer M, Posé D, Ott F, Schmid M, Carles CC. Dynamics of H3K4me3 Chromatin Marks Prevails over H3K27me3 for Gene Regulation during Flower Morphogenesis in Arabidopsis thaliana. Epigenomes. 2017; 1(2):8. https://doi.org/10.3390/epigenomes1020008
Chicago/Turabian StyleEngelhorn, Julia, Robert Blanvillain, Christian Kröner, Hugues Parrinello, Marine Rohmer, David Posé, Felix Ott, Markus Schmid, and Cristel C. Carles. 2017. "Dynamics of H3K4me3 Chromatin Marks Prevails over H3K27me3 for Gene Regulation during Flower Morphogenesis in Arabidopsis thaliana" Epigenomes 1, no. 2: 8. https://doi.org/10.3390/epigenomes1020008
APA StyleEngelhorn, J., Blanvillain, R., Kröner, C., Parrinello, H., Rohmer, M., Posé, D., Ott, F., Schmid, M., & Carles, C. C. (2017). Dynamics of H3K4me3 Chromatin Marks Prevails over H3K27me3 for Gene Regulation during Flower Morphogenesis in Arabidopsis thaliana. Epigenomes, 1(2), 8. https://doi.org/10.3390/epigenomes1020008