
Adaptive Evolution and Transcriptomic Specialization of P450 Detoxification Genes in the Colorado Potato Beetle Across Developmental Stages and Tissues


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Transcriptome Analysis
2.3. Identification of Stage- and Tissue-Specific Genes
2.4. Gene Functional Analysis
2.5. Identification and Location of P450 Genes
2.6. Positive Selective Pressure Analysis
2.7. Genetic Variant Detection
3. Results
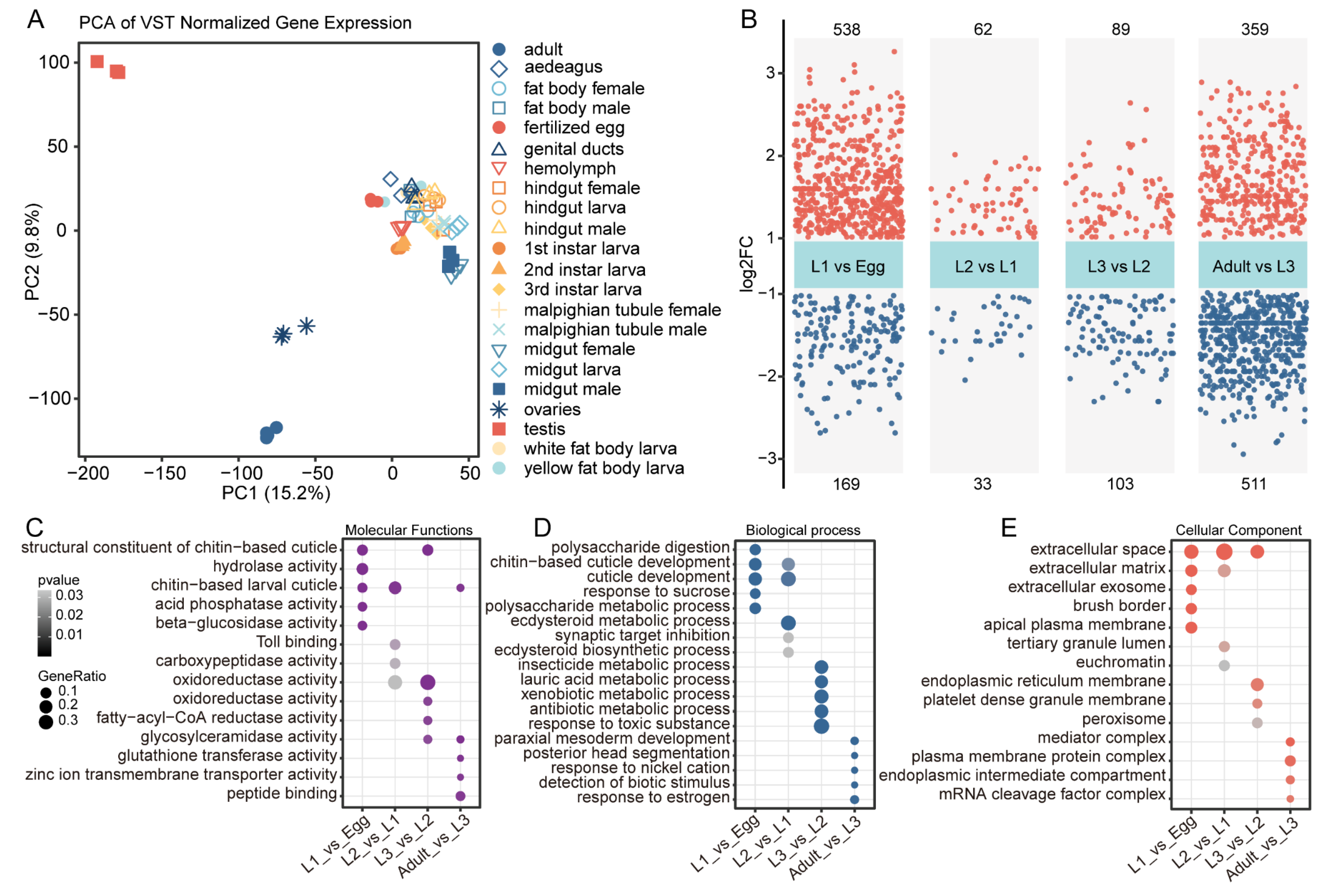
3.1. Differential Expression Analysis Across Sequential Developmental Stages
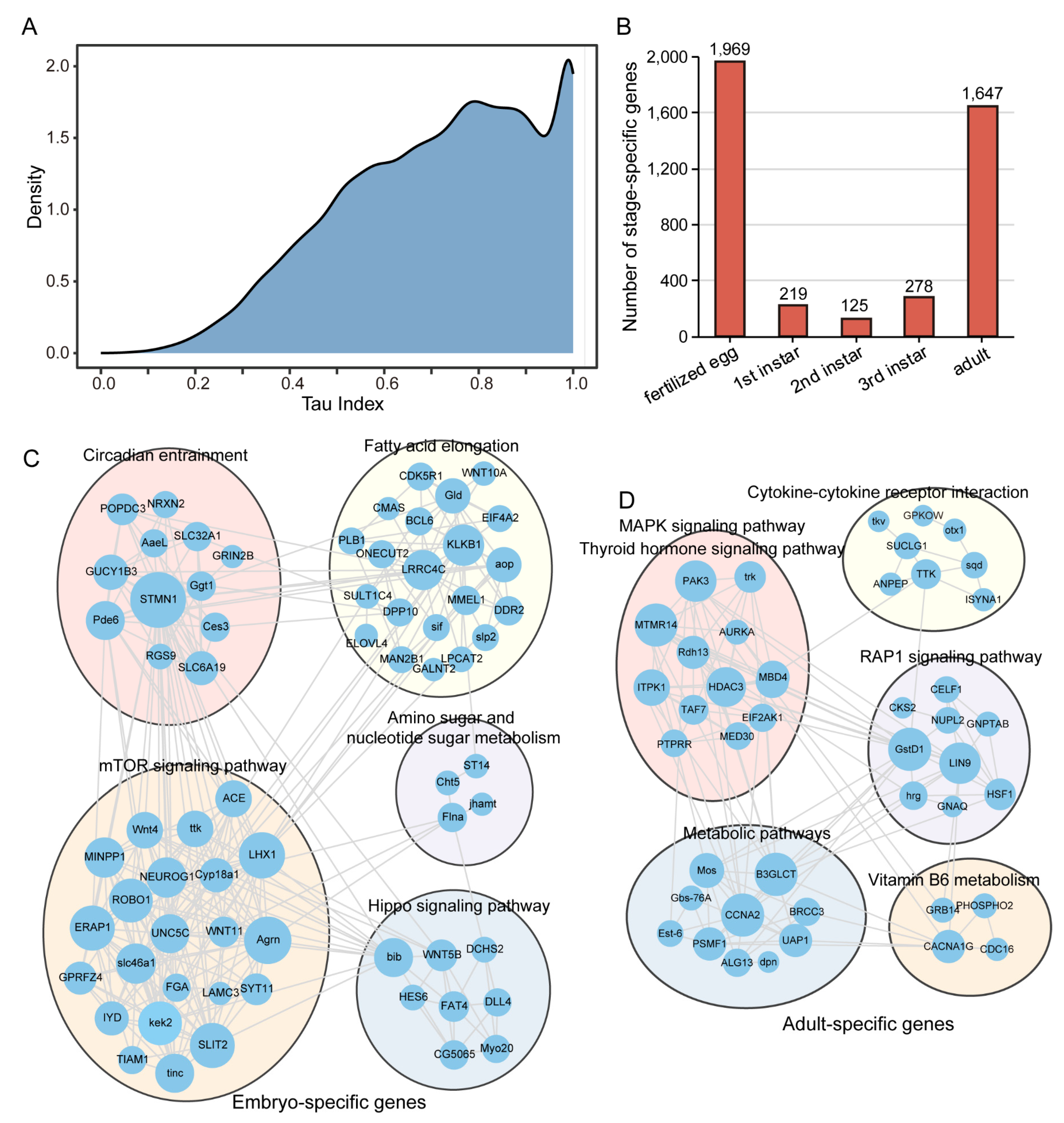
3.2. Stage-Specific Gene Expression Patterns in Colorado Potato Beetle Development
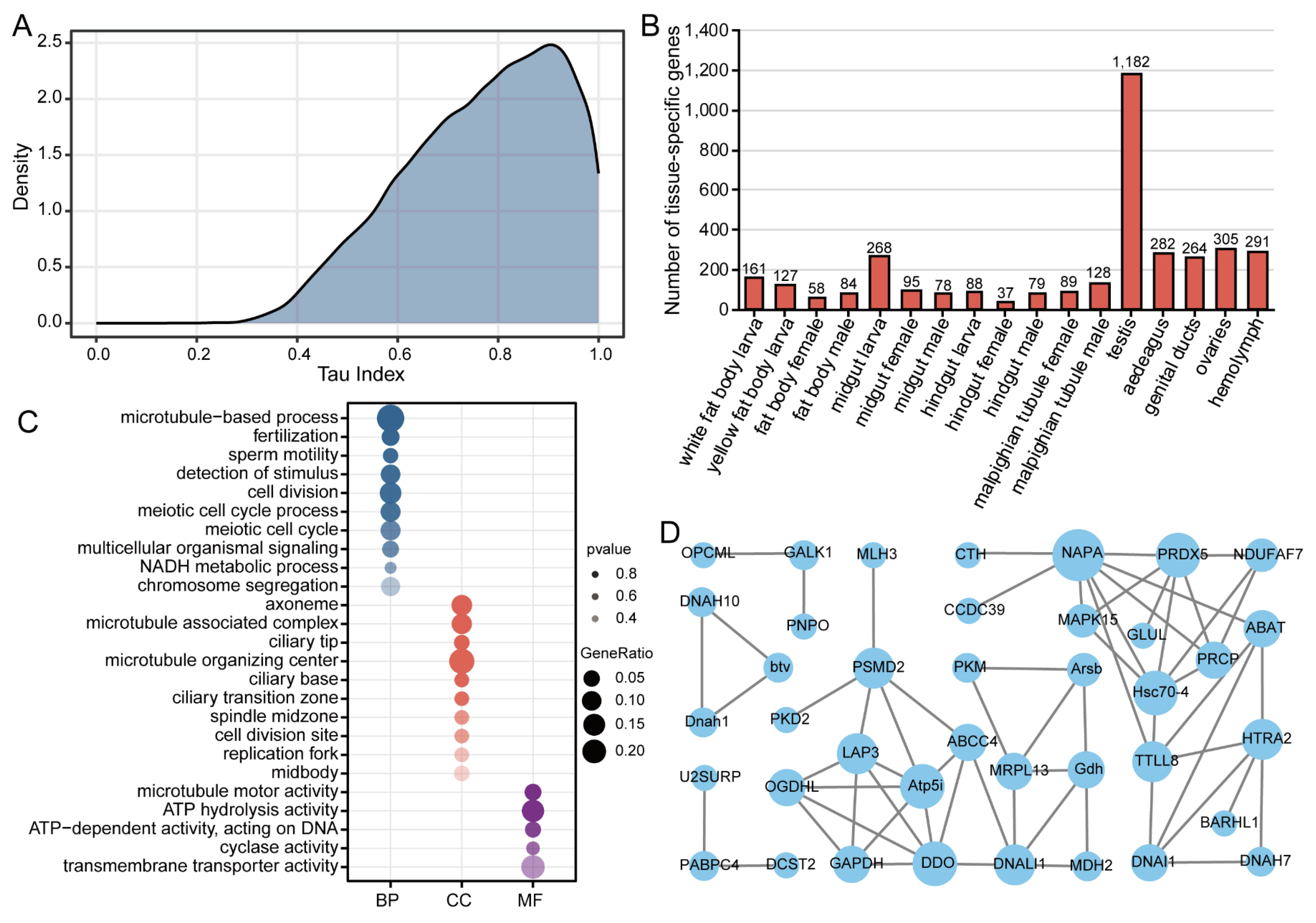
3.3. Identification and Functional Characterization of Tissue-Specific Genes
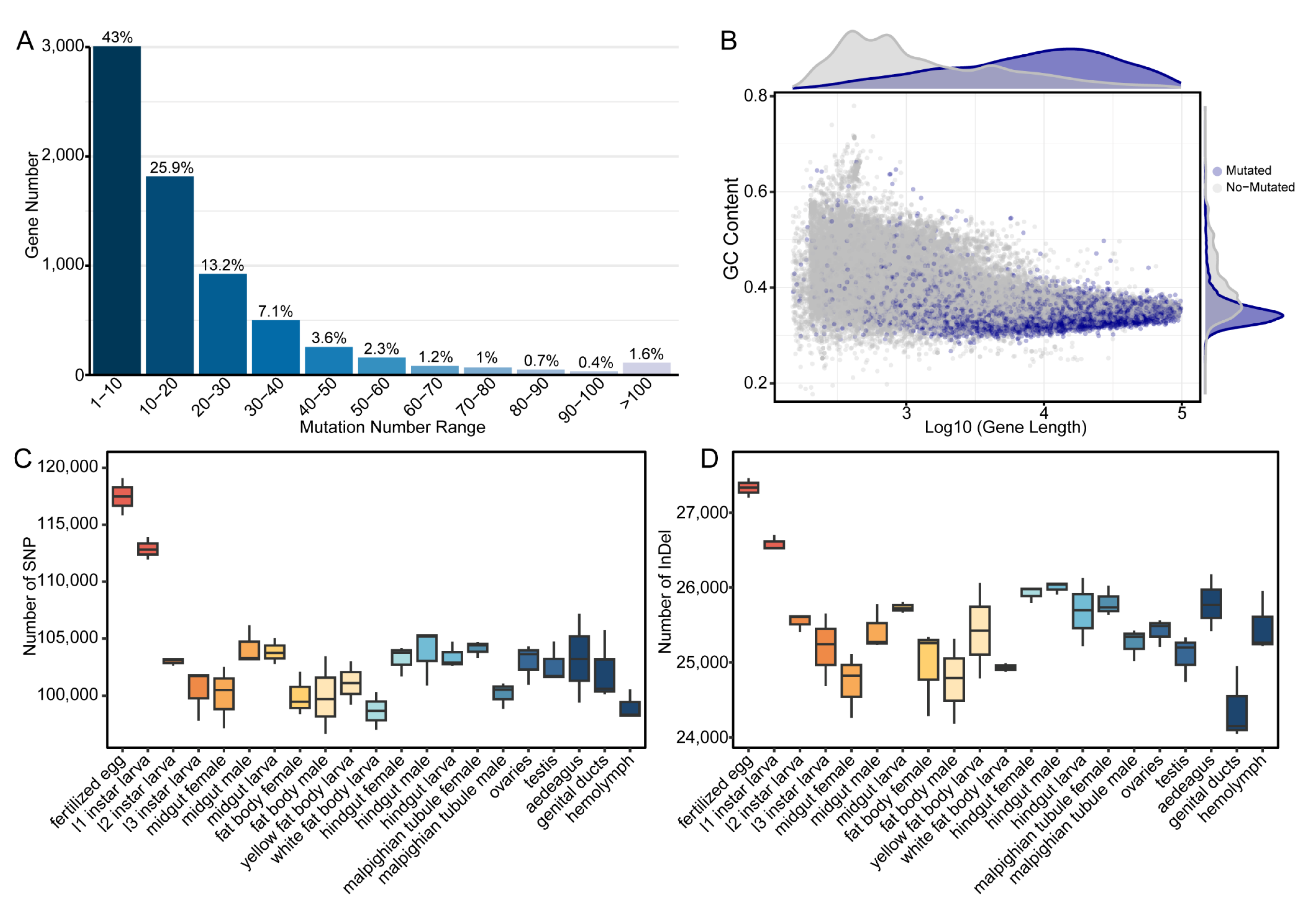
3.4. Analysis of mRNA Sequence Polymorphisms Across Developmental Stages and Tissues
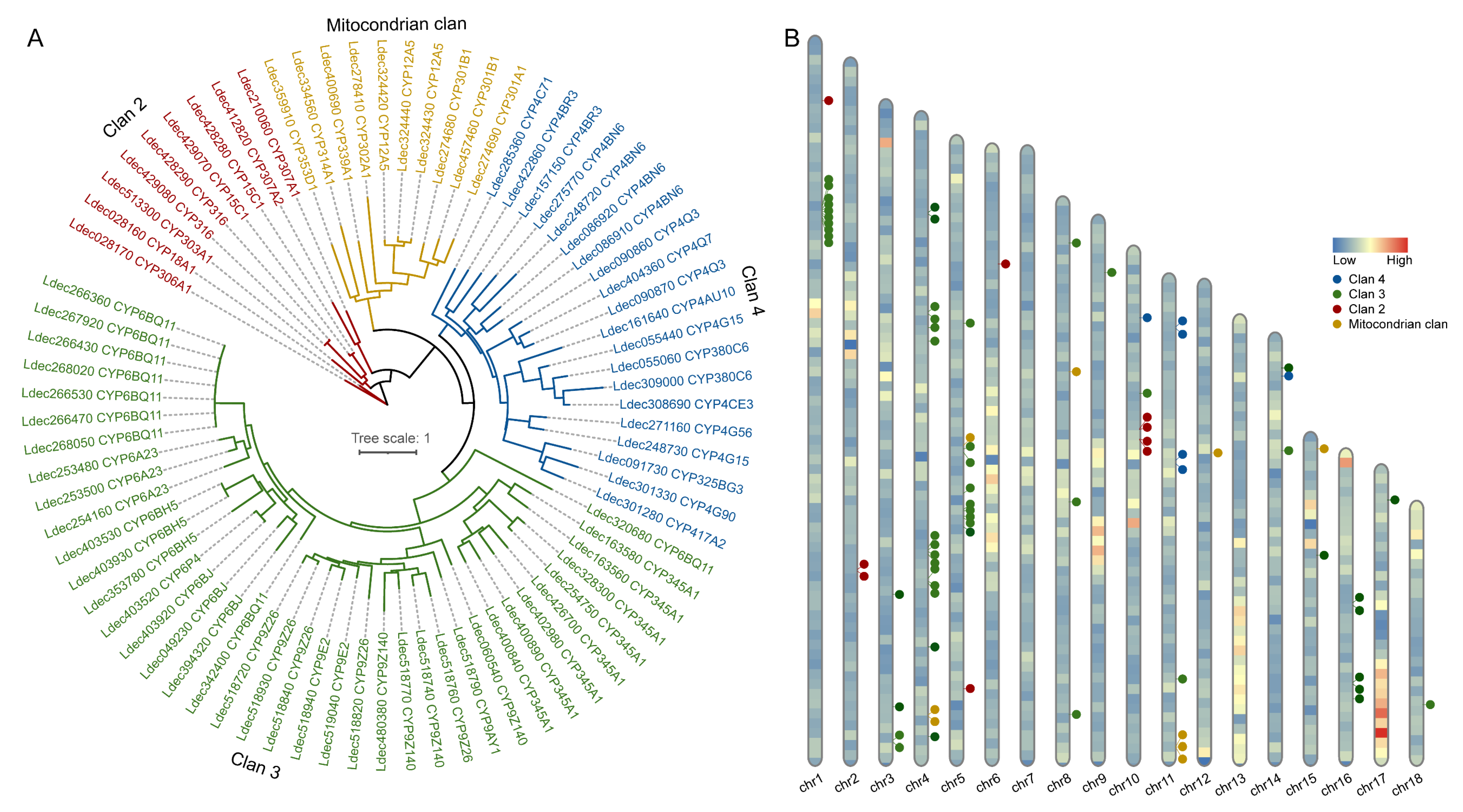
3.5. Identification of P450 Genes in Colorado Potato Beetle
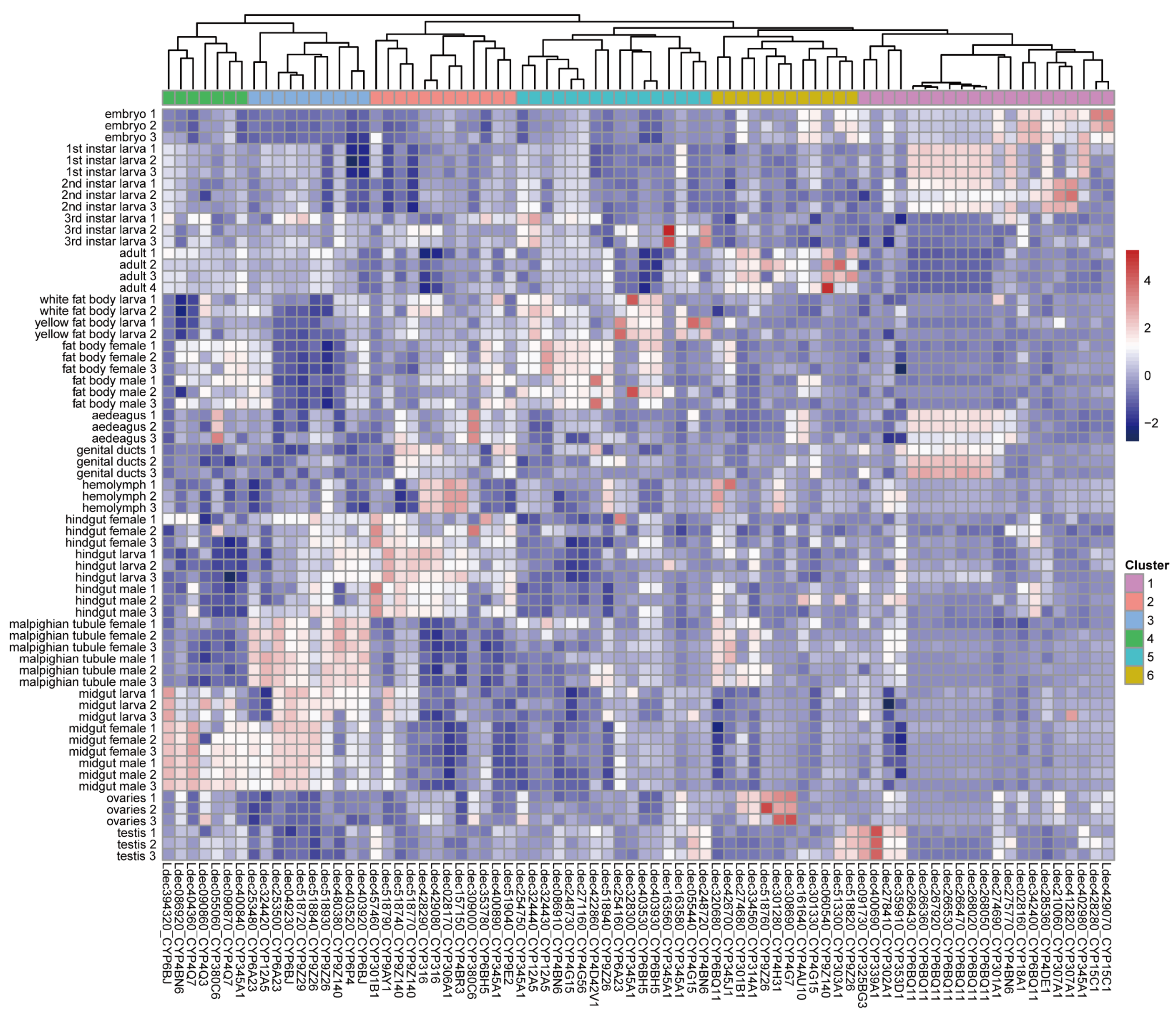
3.6. Expression Profiles of P450 Genes in Colorado Potato Beetle
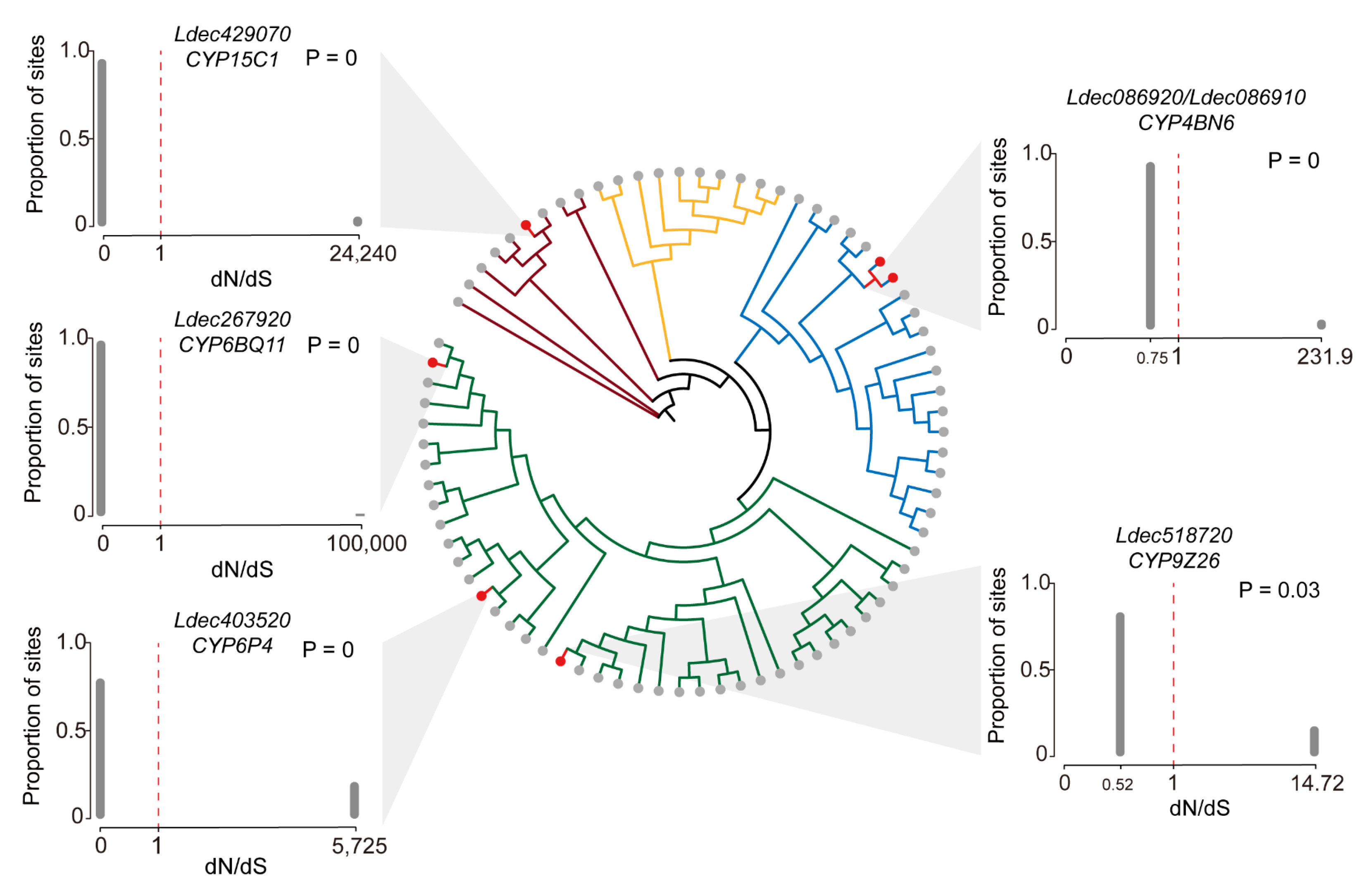
3.7. Evolutionary Selection Pressure of P450 Genes in Colorado Potato Beetle
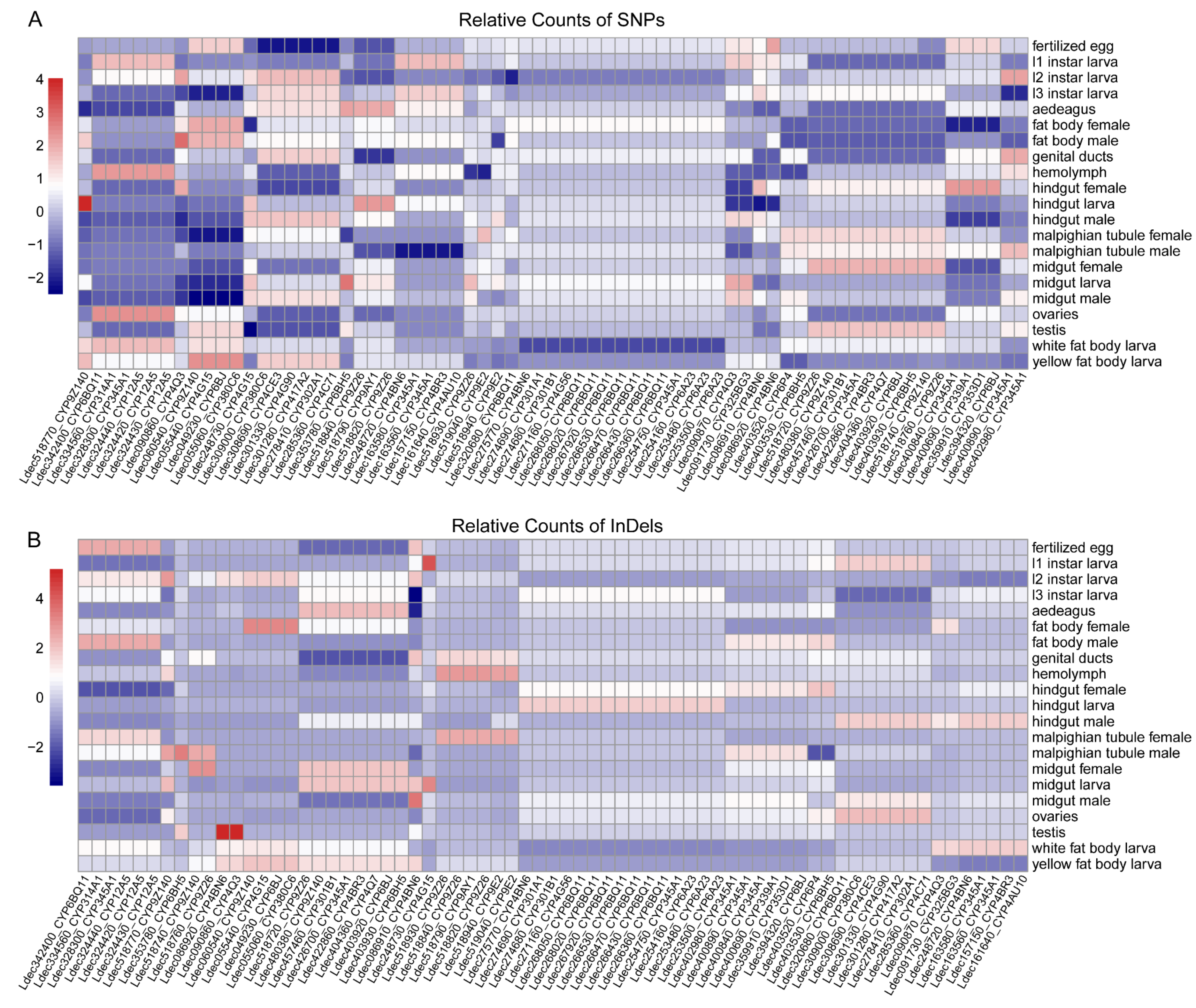
3.8. Tissue and Developmental Stage-Specific Mutation Patterns in P450 mRNA Transcripts
4. Discussion
4.1. Differential Expression Analysis Reveals Gene Expression Dynamics During Key Developmental Transitions in Colorado Potato Beetle
4.2. Stage-Specific Gene Expression Patterns Reveal Distinct Developmental Specialization
4.3. Tissue-Specific Gene Expression Reveals Transcriptomic Specialization in Reproductive Tissues
4.4. Developmental Stage Influences Transcriptome Sequence Polymorphism
4.5. Evolutionary Selection Pressure of P450 Genes
4.6. Tissue and Developmental Stage-Specific Mutation Patterns
4.7. Limitations of RNA-Based Mutation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoville, S.D.; Chen, Y.H.; Andersson, M.N.; Benoit, J.B.; Bhandari, A.; Bowsher, J.H.; Brevik, K.; Cappelle, K.; Chen, M.-J.M.; Childers, A.K.; et al. A Model Species for Agricultural Pest Genomics: The Genome of the Colorado Potato Beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Sci. Rep. 2018, 8, 1931. [Google Scholar] [CrossRef] [PubMed]
- Grapputo, A.; Boman, S.; Lindström, L.; Lyytinen, A.; Mappes, J. The Voyage of an Invasive Species across Continents: Genetic Diversity of North American and European Colorado Potato Beetle Populations. Mol. Ecol. 2005, 14, 4207–4219. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, Y.; Zhang, R. Invasion of Colorado Potato Beetle, Leptinotarsa decemlineata, in China: Dispersal, Occurrence, and Economic Impact. Entomol. Exp. Appl. 2012, 143, 207–217. [Google Scholar] [CrossRef]
- Dai, T.-M.; Tian, H.; Liu, X.; Zhang, G.-F.; Wang, Y.-S. The Complete Mitochondrial Genome of Invasive Insect Leptinotarsa decemlineata Say 1824 (Coleoptera: Chrysomelidae). Mitochondrial DNA B Resour. 2022, 7, 358–360. [Google Scholar] [CrossRef]
- Chen, Y.H.; Cohen, Z.P.; Bueno, E.M.; Christensen, B.M.; Schoville, S.D. Rapid Evolution of Insecticide Resistance in the Colorado Potato Beetle, Leptinotarsa decemlineata. Curr. Opin. Insect Sci. 2023, 55, 101000. [Google Scholar] [CrossRef]
- Kaplanoglu, E.; Chapman, P.; Scott, I.M.; Donly, C. Overexpression of a Cytochrome P450 and a UDP-Glycosyltransferase Is Associated with Imidacloprid Resistance in the Colorado Potato Beetle, Leptinotarsa decemlineata. Sci. Rep. 2017, 7, 1762. [Google Scholar] [CrossRef]
- Clements, J.; Schoville, S.; Clements, A.; Amezian, D.; Davis, T.; Sanchez-Sedillo, B.; Bradfield, C.; Huseth, A.S.; Groves, R.L. Agricultural Fungicides Inadvertently Influence the Fitness of Colorado Potato Beetles, Leptinotarsa decemlineata, and Their Susceptibility to Insecticides. Sci. Rep. 2018, 8, 13282. [Google Scholar] [CrossRef]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene Expression across Mammalian Organ Development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Feyereisen, R. Evolution of Insect P450. Biochem. Soc. Trans. 2006, 34, 1252–1255. [Google Scholar] [CrossRef]
- Li, Y.; Drummond, A.; Sawayama, A.; Snow, C.; Bloom, J.; Arnold, F. A Diverse Family of Thermostable Cytochrome P450s Created by Recombination of Stabilizing Fragments. Nat. Biotechnol. 2007, 25, 1051–1056. [Google Scholar] [CrossRef]
- Liu, N.; Li, M.; Gong, Y.; Liu, F.; Li, T. Cytochrome P450s—Their Expression, Regulation, and Role in Insecticide Resistance. Pestic. Biochem. Phys. 2015, 120, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Mokhosoev, I.; Astakhov, D.; Terentiev, A.; Moldogazieva, N. Cytochrome P450 Monooxygenase Systems: Diversity and Plasticity for Adaptive Stress Response. Prog. Biophys. Mol. Biol. 2024, 193, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Pottier, M.-A.; Bozzolan, F.; Chertemps, T.; Jacquin-Joly, E.; Lalouette, L.; Siaussat, D.; Maïbèche-Coisne, M. Cytochrome P450s and Cytochrome P450 Reductase in the Olfactory Organ of the Cotton Leafworm Spodoptera littoralis. Insect Mol. Biol. 2012, 21, 568–580. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Li, L.; Song, Q.; Stanley, D.; Wei, S.; Zhu, J. Genome-Wide and Expression-Profiling Analyses of the Cytochrome P450 Genes in Tenebrionidea. Arch. Insect Biochem. Physiol. 2022, 111, e21954. [Google Scholar] [CrossRef]
- Lu, K.; Song, Y.; Zeng, R. The Role of Cytochrome P450-Mediated Detoxification in Insect Adaptation to Xenobiotics. Curr. Opin. Insect Sci. 2021, 43, 103–107. [Google Scholar] [CrossRef]
- Nauen, R.; Bass, C.; Feyereisen, R.; Vontas, J. The Role of Cytochrome P450s in Insect Toxicology and Resistance. Annu. Rev. Entomol. 2021, 67, 105–124. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, Y.; Zhou, D.; Fang, J.; Cao, J.; Shi, C.; Lei, Y.; Fu, K.; Guo, W.; Jiang, W. Expression and Functional Analysis of Two Cytochrome P450 Monooxygenase Genes and a UDP-Glycosyltransferase Gene Linked with Thiamethoxam Resistance in the Colorado Potato Beetle. Insects 2024, 15, 559. [Google Scholar] [CrossRef]
- Wang, D.-Z.; Jiang, Z.; Shen, Z.; Wang, H.; Wang, B.; Shou, W.; Zheng, H.; Chu, X.; Shi, J.; Huang, W. Functional Evaluation of Genetic and Environmental Regulators of P450 mRNA Levels. PLoS ONE 2011, 6, e24900. [Google Scholar] [CrossRef]
- Schuler, M.; Berenbaum, M. Structure and Function of Cytochrome P450S in Insect Adaptation to Natural and Synthetic Toxins: Insights Gained from Molecular Modeling. J. Chem. Ecol. 2013, 39, 1232–1245. [Google Scholar] [CrossRef]
- Wan, P.; Shi, X.; Kong, Y.; Zhou, L.-T.; Guo, W.; Ahmat, T.; Li, G.-Q. Identification of Cytochrome P450 Monooxygenase Genes and Their Expression Profiles in Cyhalothrin-Treated Colorado Potato Beetle, Leptinotarsa decemlineata. Pestic. Biochem. Phys. 2013, 107, 360–368. [Google Scholar] [CrossRef]
- Sayers, E.; Bolton, E.; Brister, J.; Canese, K.; Chan, J.; Comeau, D.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, L.; Wang, Y.; Xu, S. Gene Expression Atlas of the Colorado Potato Beetle (Leptinotarsa decemlineata). Sci. Data 2025, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, C.; Zhang, M.; Zhou, H.; Zuo, Z.; Ding, X.; Zhang, R.; Li, F.; Gao, Y. Chromosome-Level Genome Assembly of the Colorado Potato Beetle, Leptinotarsa decemlineata. Sci. Data 2023, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A Benchmark of Gene Expression Tissue-Specificity Metrics. Brief Bioinform. 2015, 18, 205–214. [Google Scholar] [CrossRef]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-Wide Midrange Transcription Profiles Reveal Expression Level Relationships in Human Tissue Specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.; Giner-Lamia, J.; Mende, D.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.; et al. eggNOG 6.0: Enabling Comparative Genomics across 12,535 Organisms. Nucleic Acids Res. 2022, 51, D389–D394. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cheng, Z.; Chen, L.; Yan, B.; Zhang, M.; Tian, J.; Mei, Y. Insect-eP450DB: A Resource of Functionally Characterized P450s in Insects. Insect Sci. 2025. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.; Schmidt, H.; Chernomor, O.; Schrempf, D.; Woodhams, M.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2019, 37, 1530–1534. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust Conversion of Protein Sequence Alignments into the Corresponding Codon Alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Wertheim, J.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.K. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection Permalink. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.K.; Poon, A.; Velazquez, R.; Weaver, S.; Hepler, N.; Murrell, B.; Shank, S.; Magalis, B.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2019, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Davis, S. Vcftoolz: A Python Package for Comparing and Evaluating Variant Call Format Files. J. Open Source Softw. 2019, 4, 1144. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.; Whitwham, A.; Keane, T.; McCarthy, S.; Davies, R.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2020, 10, giab008. [Google Scholar] [CrossRef]
- Zhou, H.; Shu, R.; Zhang, C.; Xiao, Y.; Jing, D.; Tang, J.; Cao, Z.; Chen, X.; Mei, Y.; Li, F. Developmental Correspondence of Juvenile Stages across the Locust, Harlequin Ladybird, and Diamondback Moth. iScience 2024, 27, 110898. [Google Scholar] [CrossRef]
- Merzendorfer, H.; Zimoch, L. Chitin Metabolism in Insects: Structure, Function and Regulation of Chitin Synthases and Chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef]
- Graveley, B. The Developmental Transcriptome of Drosophila melanogaster. Genome Biol. 2011, 11, I11. [Google Scholar] [CrossRef]
- Wu, Z.; Guan, K. Hippo Signaling in Embryogenesis and Development. Trends Biochem. Sci. 2021, 46, 51–63. [Google Scholar] [CrossRef]
- Correia, B.; Sousa, M.; Ramalho-Santos, J. The mTOR Pathway in Reproduction: From Gonadal Function to Developmental Coordination. Reproduction 2020, 159, R173–R188. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Hou, L.; Wang, X.; Kang, L. Core Transcriptional Signatures of Phase Change in the Migratory Locust. Protein Cell 2019, 10, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, Y.; Cheng, Y.; Tian, S.; Bai, Y.; Zuo, J.; Palli, S.; Chen, X. Knockout of a Testis-Specific Gene Cluster Impairs Male Fertility in the Fall Armyworm, Spodoptera frugiperda. Pest Manag. Sci. 2025, 81, 2355–2363. [Google Scholar] [CrossRef]
- Lau, A.; Xu, Y.-M. Regulation of Human Mitogen-activated Protein Kinase 15 (Extracellular Signal-regulated Kinase 7/8) and Its Functions: A Recent Update. J. Cell Physiol. 2018, 234, 75–88. [Google Scholar] [CrossRef]
- Gudkov, M.; Thibaut, L.; Giannoulatou, E. Quantifying Negative Selection on Synonymous Variants. HGG Adv. 2024, 5, 100262. [Google Scholar] [CrossRef]
- Fu, Q.; Zhou, J.; Luan, S.; Dai, P.; Lyu, D.; Chen, B.; Luo, K.; Kong, J.; Meng, X. Analysis of Elimination Effects of Inbreeding on Genotype Frequency in Larval Stages of Chinese Shrimp. Biology 2024, 13, 268. [Google Scholar] [CrossRef]
- Cannavó, E.; Koelling, N.; Harnett, D.; Garfield, D.; Casale, F.; Ciglar, L.; Gustafson, H.; Viales, R.; Marco-Ferreres, R.; Degner, J.; et al. Genetic Variants Regulating Expression Levels and Isoform Diversity during Embryogenesis. Nature 2016, 541, 402–406. [Google Scholar] [CrossRef]
- Peregrín-Alvarez, J.; Sanford, C.; Parkinson, J. The Conservation and Evolutionary Modularity of Metabolism. Genome Biol. 2009, 10, R63. [Google Scholar] [CrossRef]
- Feyereisen, R. Arthropod CYPomes Illustrate the Tempo and Mode in P450 Evolution. Biochim. Biophys. Acta 2011, 1814, 19–28. [Google Scholar] [CrossRef]
- Ian, P.R.; Elizabeth, S.A.; Nelson, D.R. Cytochrome P450 Nomenclature, 2004. Methods Mol. Biol. 2006, 320, 1–10. [Google Scholar] [CrossRef]
- Helvig, C.; Koener, J.F.; Unnithan, G.C.; Feyereisen, R. CYP15A1, the Cytochrome P450 That Catalyzes Epoxidation of Methyl Farnesoate to Juvenile Hormone III in Cockroach Corpora Allata. Proc. Natl. Acad. Sci. USA 2004, 101, 4024–4029. [Google Scholar] [CrossRef] [PubMed]
- Jindra, M.; Palli, S.R.; Riddiford, L.M. The Juvenile Hormone Signaling Pathway in Insect Development. Annu. Rev. Entomol. 2013, 58, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Palli, S.R. Molecular Analysis of Nutritional and Hormonal Regulation of Female Reproduction in the Red Flour Beetle, Tribolium castaneum. Insect Mol. Biol. 2011, 41, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, D.L. Regulation of Diapause. Annu. Rev. Entomol. 2002, 47, 93–122. [Google Scholar] [CrossRef]
- Koštál, V. Eco-Physiological Phases of Insect Diapause. J. Insect Physiol. 2006, 52, 113–127. [Google Scholar] [CrossRef]
- Scott, J.G.; Liu, N.; Wen, Z. Insect Cytochromes P450: Diversity, Insecticide Resistance and Tolerance to Plant Toxins. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1998, 121, 147–155. [Google Scholar] [CrossRef]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular Mechanisms of Metabolic Resistance to Synthetic and Natural Xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef]
- Daborn, P.J.; Yen, J.L.; Bogwitz, M.R.; Le Goff, G.; Feil, E.; Jeffers, S.; Tijet, N.; Perry, T.; Heckel, D.; Batterham, P.; et al. A Single P450 Allele Associated with Insecticide Resistance in Drosophila. Science 2002, 297, 2253–2256. [Google Scholar] [CrossRef]
- Müller, P.; Warr, E.; Stevenson, B.J.; Pignatelli, P.M.; Morgan, J.C.; Steven, A.; Yawson, A.E.; Mitchell, S.N.; Ranson, H.; Hemingway, J.; et al. Field-Caught Permethrin-Resistant Anopheles gambiae Overexpress CYP6P3, a P450 That Metabolises Pyrethroids. PLoS Genet. 2008, 4, e1000286. [Google Scholar] [CrossRef]
- Stevenson, B.J.; Bibby, J.; Pignatelli, P.; Muangnoicharoen, S.; O’Neill, P.M.; Lian, L.-Y.; Müller, P.; Nikou, D.; Steven, A.; Hemingway, J.; et al. Cytochrome P450 6M2 from the Malaria Vector Anopheles Gambiae Metabolizes Pyrethroids: Sequential Metabolism of Deltamethrin Revealed. Insect Biochem. Mol. Biol. 2011, 41, 492–502. [Google Scholar] [CrossRef]
- Zhu, F.; Moural, T.W.; Nelson, D.R.; Palli, S.R. A Specialist Herbivore Pest Adaptation to Xenobiotics through Up-Regulation of Multiple Cytochrome P450s. Sci. Rep. 2016, 6, 20421. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A. The Role of Cytochrome P450 Monooxygenases in Plant-Insect Interactions. Plant Physiol. 1996, 112, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Potato Glycoalkaloids and Metabolites: Roles in the Plant and in the Diet. J. Agric. Food Chem. 2006, 54, 8655–8681. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.R.; Vanderwel, D.; Choi, S.; Pomonis, J.G.; Reitz, R.C.; Blomquist, G.J. Unusual Mechanism of Hydrocarbon Formation in the Housefly: Cytochrome P450 Converts Aldehyde to the Sex Pheromone Component (Z)-9-Tricosene and CO2. Proc. Natl. Acad. Sci. USA 1994, 91, 10000–10004. [Google Scholar] [CrossRef]
- Xiong, W.; Gao, S.; Mao, J.; Wei, L.; Xie, J.; Liu, J.; Bi, J.; Song, X.; Li, B. CYP4BN6 and CYP6BQ11 Mediate Insecticide Susceptibility and Their Expression Is Regulated by Latrophilin in Tribolium castaneum. Pest Manag. Sci. 2019, 75, 2744–2755. [Google Scholar] [CrossRef]
- Howard, R.W.; Blomquist, G.J. Ecological, Behavioral, and Biochemical Aspects of Insect Hydrocarbons. Annu. Rev. Entomol. 2005, 50, 371–393. [Google Scholar] [CrossRef]
- Gould, F. Sustainability of Transgenic Insecticidal Cultivars: Integrating Pest Genetics and Ecology. Annu. Rev. Entomol. 1998, 43, 701–726. [Google Scholar] [CrossRef]
- Rabani, M.; Raychowdhury, R.; Jovanović, M.; Rooney, M.; Stumpo, D.; Pauli, A.; Hacohen, N.; Schier, A.; Blackshear, P.; Friedman, N.; et al. High-Resolution Sequencing and Modeling Identifies Distinct Dynamic RNA Regulatory Strategies. Cell 2014, 159, 1698–1710. [Google Scholar] [CrossRef]
- Flickinger, R. Polymorphism of Simple Sequence Repeats May Quantitatively Regulate Gene Transcription. Exp. Cell Res. 2020, 390, 111969. [Google Scholar] [CrossRef]
- Furey, T.S.; Diekhans, M.; Lu, Y.; Graves, T.A.; Oddy, L.; Randall-Maher, J.; Hillier, L.W.; Wilson, R.K.; Haussler, D. Analysis of Human mRNAs With the Reference Genome Sequence Reveals Potential Errors, Polymorphisms, and RNA Editing. Genome Res. 2004, 14, 2034–2040. [Google Scholar] [CrossRef]
- Shen, L.X.; Basilion, J.P.; Stanton, V.P. Single-Nucleotide Polymorphisms Can Cause Different Structural Folds of mRNA. Proc. Natl. Acad. Sci. USA 1999, 96, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lv, J.; Gui, B.; Yin, H.; Wu, X.; Zhang, Y.; Jin, Y. A-to-I RNA Editing Alters Less-Conserved Residues of Highly Conserved Coding Regions: Implications for Dual Functions in Evolution. RNA 2008, 14, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, K.R.; Coolon, J.D.; Wittkopp, P.J. Sources of Bias in Measures of Allele-Specific Expression Derived from RNA-Seq Data Aligned to a Single Reference Genome. BMC Genom. 2013, 14, 536. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome Variation in Human Tissues Revealed by Long-Read Sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef]
- Pelechano, V.; Wei, W.; Steinmetz, L.M. Extensive Transcriptional Heterogeneity Revealed by Isoform Profiling. Nature 2013, 497, 127–131. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Cheng, Z.; Tang, J.; Lu, Y.; Mei, Y.; Chen, X. Adaptive Evolution and Transcriptomic Specialization of P450 Detoxification Genes in the Colorado Potato Beetle Across Developmental Stages and Tissues. Insects 2025, 16, 608. https://doi.org/10.3390/insects16060608
Zhou H, Cheng Z, Tang J, Lu Y, Mei Y, Chen X. Adaptive Evolution and Transcriptomic Specialization of P450 Detoxification Genes in the Colorado Potato Beetle Across Developmental Stages and Tissues. Insects. 2025; 16(6):608. https://doi.org/10.3390/insects16060608
Chicago/Turabian StyleZhou, Hang, Ziqi Cheng, Jiejing Tang, Yueqi Lu, Yang Mei, and Xi Chen. 2025. "Adaptive Evolution and Transcriptomic Specialization of P450 Detoxification Genes in the Colorado Potato Beetle Across Developmental Stages and Tissues" Insects 16, no. 6: 608. https://doi.org/10.3390/insects16060608
APA StyleZhou, H., Cheng, Z., Tang, J., Lu, Y., Mei, Y., & Chen, X. (2025). Adaptive Evolution and Transcriptomic Specialization of P450 Detoxification Genes in the Colorado Potato Beetle Across Developmental Stages and Tissues. Insects, 16(6), 608. https://doi.org/10.3390/insects16060608