Genomic Analysis Reveals the Role of New Genes in Venom Regulatory Network of Parasitoid Wasps

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Phylogenetic Analysis
2.3. Dating the Protein-Coding Genes Within Parasitoid Wasps
2.4. Selection Analysis
2.5. Expression Pattern of New Genes
2.6. Co-Expression Network Analysis
2.7. Total RNA Isolation, cDNA Synthesis, and Real-Time Quantitative PCR
3. Results
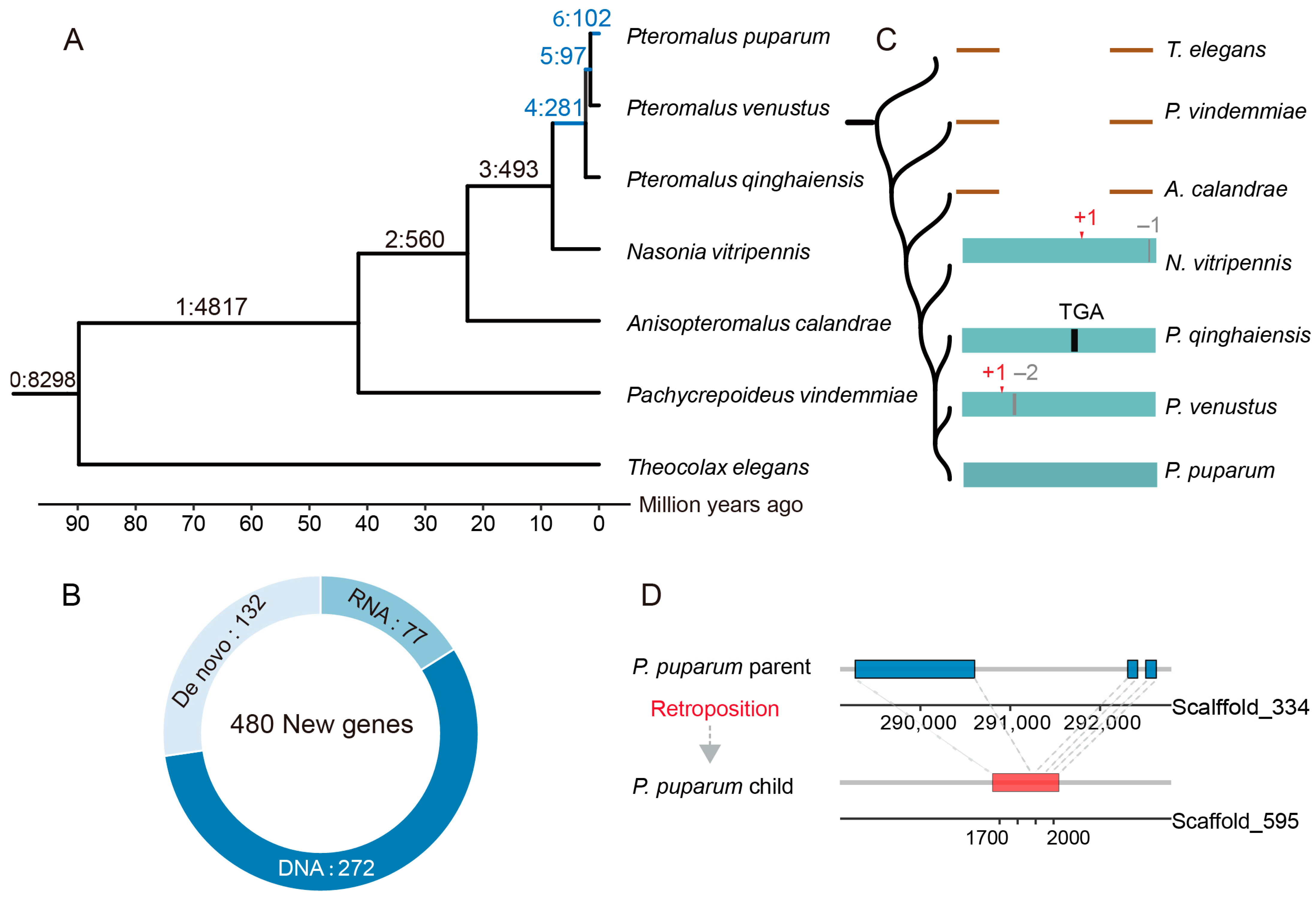
3.1. Identification and Origin of New Genes in the Parasitoid Wasps
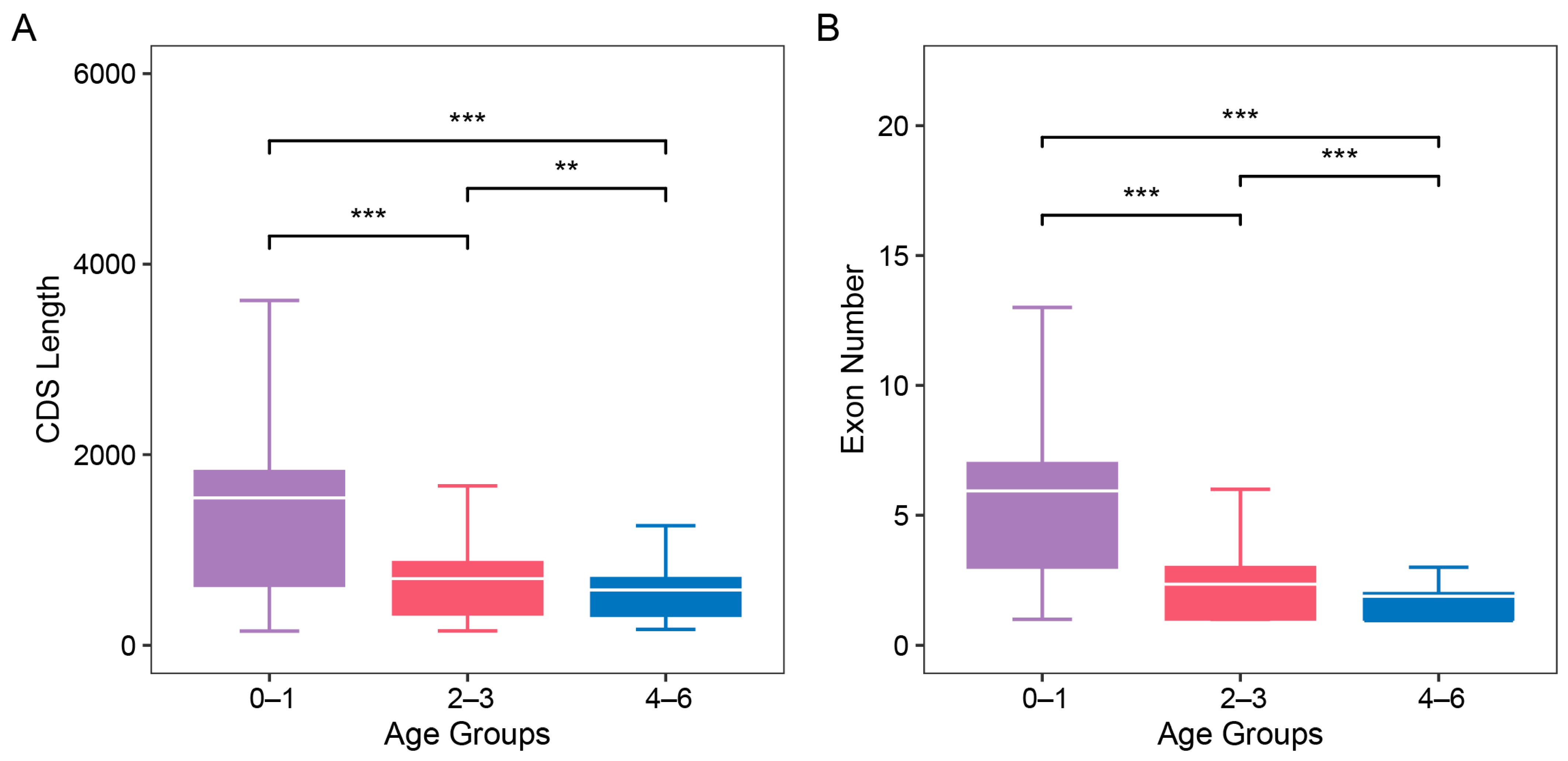
3.2. The Structural and Evolutionary Analysis of the New Genes
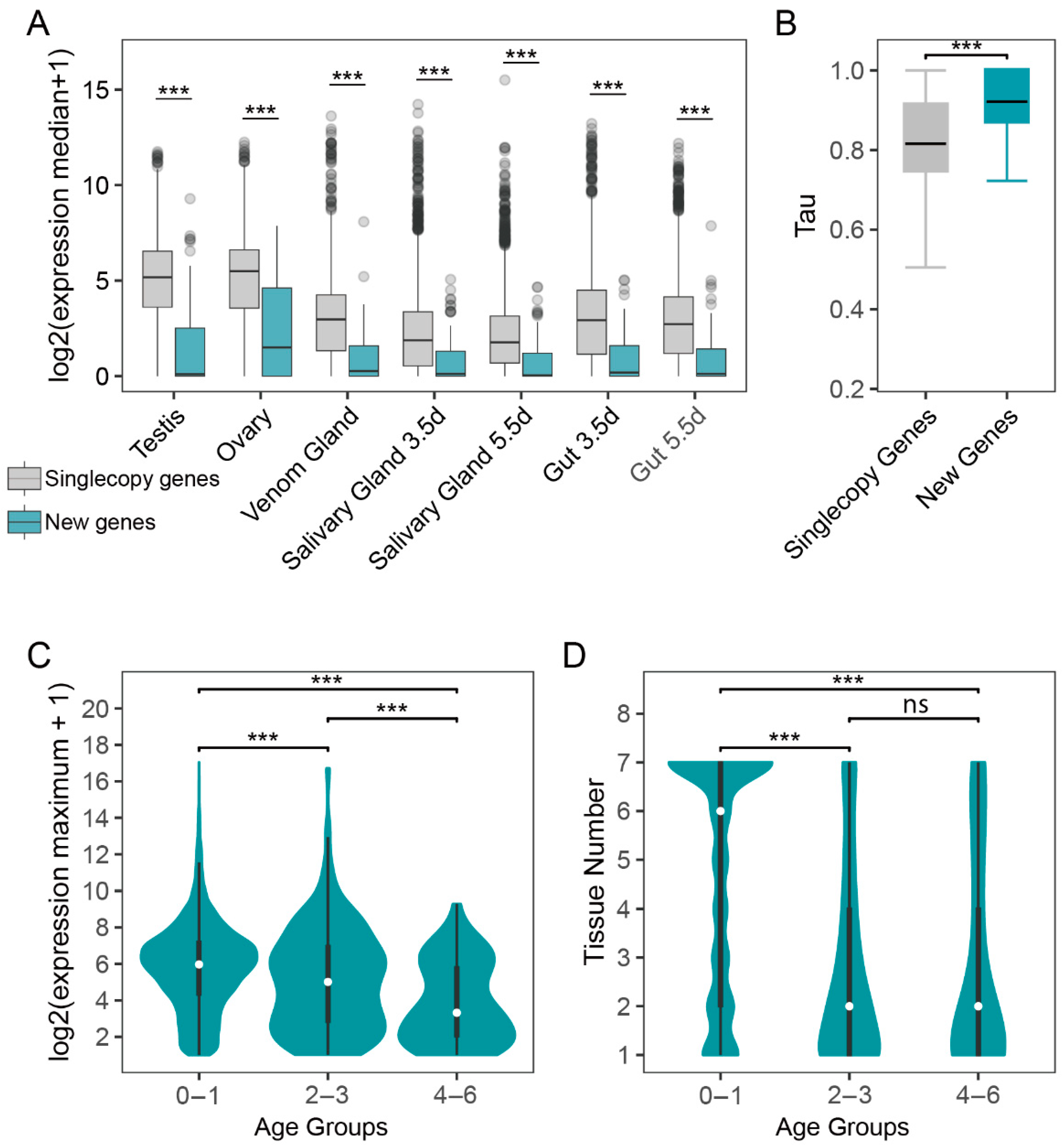
3.3. Tissue-Specific Expression Pattern of New Genes
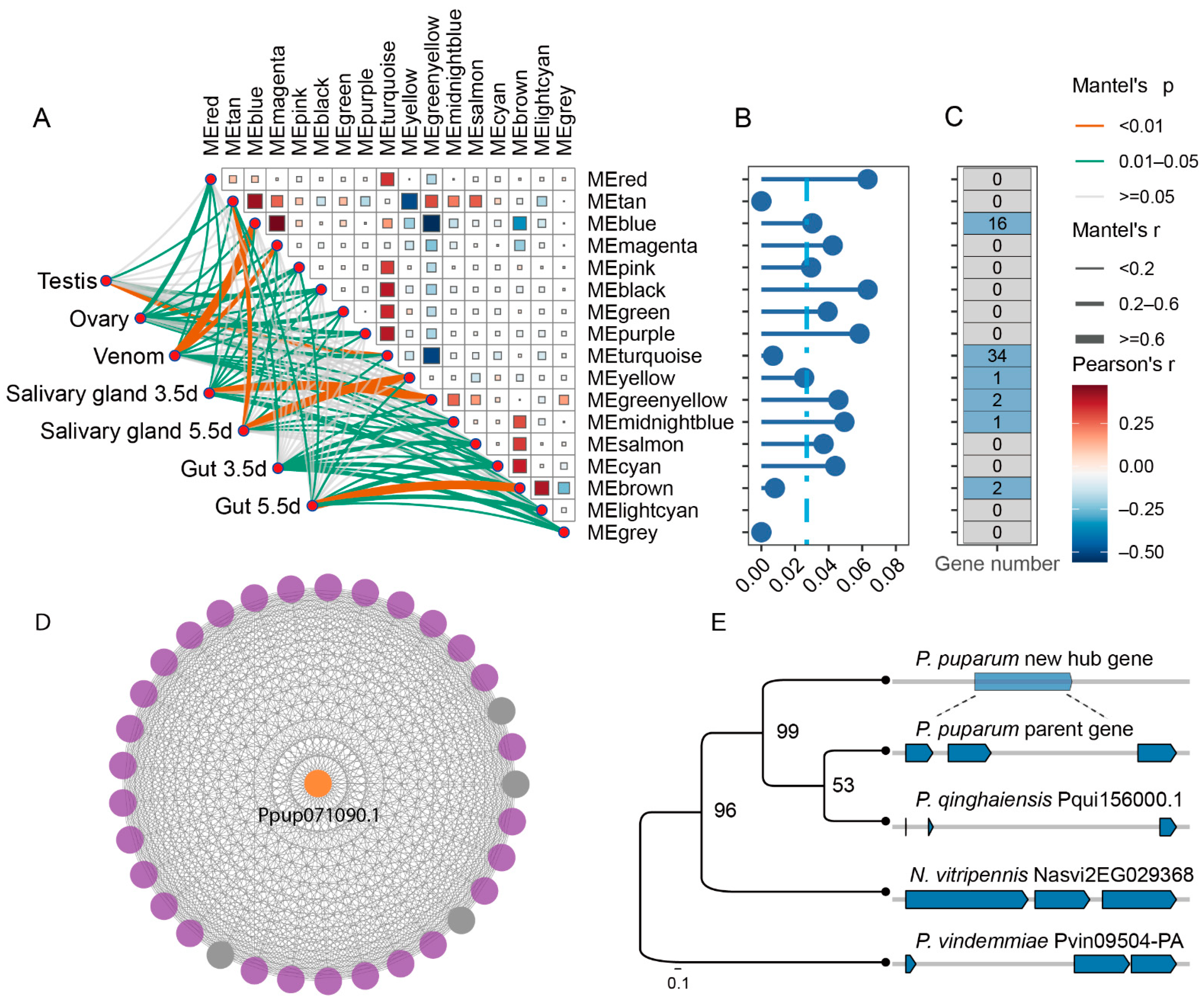
3.4. A New Gene Serves as a Hub Gene of Venom Gene Regulatory Network
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Rödelsperger, C.; Prabh, N.; Sommer, R.J. New gene origin and deep taxon phylogenomics: Opportunities and challenges. Trends Genet. 2019, 35, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Krinsky, B.H.; Long, M.Y. New genes as drivers of phenotypic evolution. Nat. Rev. Genet. 2013, 14, 645–660, Erratum in Nat. Rev. Genet. 2013, 14, 744. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Yang, T.; Li, G.W.; Chen, J.H.; Gschwend, A.R.; Yu, Y.; Hou, G.X.; Zi, J.; Zhou, R.; et al. Rapid evolution of protein diversity by de novo origination in Oryza. Nat. Ecol. Evol. 2019, 3, 679–690. [Google Scholar] [CrossRef]
- Shao, Y.; Chen, C.Y.; Shen, H.; He, B.Z.; Yu, D.Q.; Jiang, S.; Zhao, S.L.; Gao, Z.Q.; Zhu, Z.L.; Chen, X.; et al. GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res. 2019, 29, 682–696. [Google Scholar] [CrossRef]
- Li, H.R.; Chen, C.Y.; Wang, Z.K.; Wang, K.; Li, Y.X. Wang W. Pattern of new gene origination in a special fish lineage, the flatfishes. Genes 2021, 12, 1819. [Google Scholar] [CrossRef]
- An, N.A.; Zhang, J.; Mo, F.; Luan, X.K.; Tian, L.; Shen, Q.S.; Li, X.S.; Li, C.Q.; Zhou, F.Q.; Zhang, B.Y.; et al. De novo genes with an lncRNA origin encode unique human brain developmental functionality. Nat. Ecol. Evol. 2023, 7, 264–276. [Google Scholar] [CrossRef]
- Zhang, Y.E.; Long, M.Y. New genes contribute to genetic and phenotypic novelties in human evolution. Curr. Opin. Genet. Dev. 2014, 29, 90–96. [Google Scholar] [CrossRef]
- Long, M.Y.; Langley, C.H. Natural selection and the origin of Jingwei, a chimeric processed functional gene in Drosophila. Science 1993, 260, 91–95. [Google Scholar] [CrossRef]
- Rivard, E.L.; Ludwig, A.G.; Patel, P.H.; Grandchamp, A.; Arnold, S.E.; Berger, A.; Scott, E.M.; Kelly, B.J.; Mascha, G.C.; Bornberg-Bauer, E.; et al. A putative evolved gene required for spermatid chromatin condensation in Drosophila melanogaster. PLoS Genet. 2021, 17, e1009787. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhao, L.; Yang, S.A.; Jiang, Y.; Chen, Y.A.; Zhao, R.P.; Zhang, Y.; Zhang, G.J.; Dong, Y.; Yu, H.J.; et al. A young duplicate gene plays essential roles in spermatogenesis by regulating several Y-linked male fertility genes. PLoS Genet. 2010, 6, e1001255. [Google Scholar] [CrossRef]
- Dai, H.Z.; Chen, Y.; Chen, S.D.; Mao, Q.Y.; Kennedy, D.; Landback, P.; Eyre-Walker, A.; Du, W.; Long, M.Y. The evolution of courtship behaviors through the origination of a new gene in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 7478–7483. [Google Scholar] [CrossRef]
- Ross, B.D.; Rosin, L.; Thomae, A.W.; Hiatt, M.A.; Vermaak, D.; de la Cruz, A.F.A.; Imhof, A.; Mellone, B.G.; Malik, H.S. Stepwise evolution of essential centromere function in a neogene. Science 2013, 340, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zheng, Y.H.; Li, Y.Y.; Shi, L.P.; Zhang, J.; Ma, D.N.; You, M.S. An orphan gene enhances male reproductive success in Plutella xylostella. Mol. Biol. Evol. 2024, 41, msae142. [Google Scholar] [CrossRef]
- Martinson, E.O.; Siebert, A.L.; He, M.; Kelkar, Y.D.; Doucette, L.A.; Werren, J.H. Evaluating the evolution and function of the dynamic Venom Y protein in ectoparasitoid wasps. Insect Mol. Biol. 2019, 8, 499–508. [Google Scholar] [CrossRef]
- Burke, G.R.; Sharanowski, B.J. Parasitoid wasps. Curr. Biol. 2024, 34, R483–R488. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.H.; Yang, Y.; Zhao, X.X.; Fang, Q.; Ye, G.Y. The state of parasitoid wasp genomics. Trends Parasitol. 2024, 40, 914–929. [Google Scholar] [CrossRef]
- Ye, X.H.; Yang, Y.; Zhao, C.; Xiao, S.; Sun, Y.; He, C.; Xiong, S.J.; Zhao, X.X.; Zhang, B.; Lin, H.W.; et al. Genomic signatures associated with maintenance of genome stability and venom turnover in two parasitoid wasps. Nat. Commun. 2022, 13, 6417. [Google Scholar] [CrossRef]
- Martinson, E.O.; Mrinalini; Kelkar, Y.D.; Chang, C.H.; Werren, J.H. The evolution of venom by co-option of single-copy genes. Curr. Biol. 2017, 27, 2007–2013. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Peters, R.S.; Krogmann, L.; Mayer, C.; Donath, A.; Gunkel, S.; Meusemann, K.; Kozlov, A.; Podsiadlowski, L.; Petersen, M.; Lanfear, R.; et al. Evolutionary history of the Hymenoptera. Curr. Biol. 2017, 27, 1013–1018. [Google Scholar] [CrossRef]
- Kirilenko, B.M.; Munegowda, C.; Osipova, E.; Jebb, D.; Sharma, V.; Blumer, M.; Morales, A.E.; Ahmed, A.W.; Kontopoulos, D.G.; Hilgers, L.; et al. Integrating gene annotation with orthology inference at scale. Science 2023, 380, 368–386. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. Biodata Min. 2015, 8, 1. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wu, T.Z.; Hu, E.Q.; Xu, S.B.; Chen, M.J.; Guo, P.F.; Dai, Z.H.; Feng, T.Z.; Zhou, L.; Tang, W.L.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wright, C.J.; Smith, C.W.J.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710. [Google Scholar] [CrossRef]
- Zhang, Y.E.; Vibranovski, M.D.; Landback, P.; Marais, G.A.; Long, M. Chromosomal redistribution of male-biased genes in mammalian evolution with two bursts of gene gain on the X chromosome. PLoS Biol. 2010, 8, e1000494. [Google Scholar] [CrossRef]
- Ye, X.; He, C.; Yang, Y.; Sun, Y.H.; Xiong, S.; Chan, K.C.; Si, Y.; Xiao, S.; Zhao, X.; Lin, H.; et al. Comprehensive isoform-level analysis reveals the contribution of alternative isoforms to venom evolution and repertoire diversity. Genome Res. 2023, 33, 1554–1567. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Yang, Y.; Fang, Q.; Ye, G. Genomics of insect natural enemies in agroecosystems. Curr. Opin. Insect Sci. 2024, 68, 101298. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Yan, Z.; Yang, Y.; Xiao, S.; Chen, L.; Wang, J.; Wang, F.; Xiong, S.; Mei, Y.; Wang, F.; et al. A chromosome-level genome assembly of the parasitoid wasp Pteromalus puparum. Mol. Ecol. Resour. 2020, 20, 1384–1402. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, G.J.; Zhang, Y.; Xu, S.Y.; Zhao, R.P.; Zhan, Z.B.; Li, X.; Ding, Y.; Yang, S.A.; Wang, W. On the origin of new genes in Drosophila. Genome Res. 2008, 18, 1446–1455. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yin, Y.; Li, H.R.; Zhou, B.T.; Zhou, J.; Zhou, X.F.; Li, Z.P.; Liu, G.C.; Pan, X.Y.; Zhang, R.; et al. Ruminant-specific genes identified using high-quality genome data and their roles in rumen evolution. Sci. Bull. 2022, 67, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, E.; Taylor, J.S.; Boone, C. Retention of duplicated genes in evolution. Trends Genet. 2022, 38, 59–72. [Google Scholar] [CrossRef]
- Zheng, E.B.; Zhao, L. Protein evidence of unannotated ORFs in Drosophila reveals diversity in the evolution and properties of young proteins. eLife 2022, 11, e78772. [Google Scholar] [CrossRef]
- Vakirlis, N.; Carvunis, A.R.; McLysaght, A. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. eLlife 2020, 9, e53500. [Google Scholar] [CrossRef]
- Betrán, E.; Thornton, K.; Long, M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002, 12, 1854–1859. [Google Scholar] [CrossRef]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef]
- Guschanski, K.; Warnefors, M.; Kaessmann, H. The evolution of duplicate gene expression in mammalian organs. Genome Res. 2017, 27, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.H.; Zhao, L. The origin and structural evolution of de novo genes in Drosophila. Nat. Commun. 2024, 15, 810. [Google Scholar] [CrossRef] [PubMed]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef]
- Neme, R.; Tautz, D. Fast turnover of genome transcription across evolutionary time exposes entire non-coding DNA to gene emergence. eLife 2016, 5, e09977. [Google Scholar] [CrossRef] [PubMed]
- Witt, E.; Benjamin, S.; Svetec, N.; Zhao, L. Testis single-cell RNA-seq reveals the dynamics of de novo gene transcription and germline mutational bias in Drosophila. eLife 2019, 8, e47138. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Landback, P.; Gschwend, A.R.; Shen, B.R.; Long, M.Y. New genes drive the evolution of gene interaction networks in the human and mouse genomes. Genome Biol. 2015, 16, 202. [Google Scholar] [CrossRef]
- Zu, J.; Gu, Y.X.; Li, Y.; Li, C.T.; Zhang, W.Y.; Zhang, Y.E.; Lee, U.; Zhang, L.; Long, M.Y. Topological evolution of coexpression networks by new gene integration maintains the hierarchical and modular structures in human ancestors. Sci. China Life Sci. 2019, 62, 594–608. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Bu, Y.; Song, J.; Yuan, B.; Xiao, S.; Wang, F.; Fang, Q.; Ye, G.; Yang, Y.; Ye, X. Genomic Analysis Reveals the Role of New Genes in Venom Regulatory Network of Parasitoid Wasps. Insects 2025, 16, 502. https://doi.org/10.3390/insects16050502
Zhang B, Bu Y, Song J, Yuan B, Xiao S, Wang F, Fang Q, Ye G, Yang Y, Ye X. Genomic Analysis Reveals the Role of New Genes in Venom Regulatory Network of Parasitoid Wasps. Insects. 2025; 16(5):502. https://doi.org/10.3390/insects16050502
Chicago/Turabian StyleZhang, Bo, Yifan Bu, Jiqiang Song, Bo Yuan, Shan Xiao, Fang Wang, Qi Fang, Gongyin Ye, Yi Yang, and Xinhai Ye. 2025. "Genomic Analysis Reveals the Role of New Genes in Venom Regulatory Network of Parasitoid Wasps" Insects 16, no. 5: 502. https://doi.org/10.3390/insects16050502
APA StyleZhang, B., Bu, Y., Song, J., Yuan, B., Xiao, S., Wang, F., Fang, Q., Ye, G., Yang, Y., & Ye, X. (2025). Genomic Analysis Reveals the Role of New Genes in Venom Regulatory Network of Parasitoid Wasps. Insects, 16(5), 502. https://doi.org/10.3390/insects16050502