
Tribe Acalyptaini (Hemiptera: Tingidae: Tinginae) Revisited: Can Apomorphies in Secondary and Tertiary Structures of 18S rRNA Length-Variable Regions (LVRs) Support Tribe Validity?


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Taxa
2.2. DNA Extraction
2.3. PCR Amplification, Purification and Sequencing
2.4. Phylogenetic Analysis
2.5. Reconstruction of Secondary Structures
2.6. Prediction of Tertiary Structures
2.7. Morpho-Molecular Structures Potentially Serving as Apomorphies
3. Results
3.1. Sequence Analysis and Tree Topology
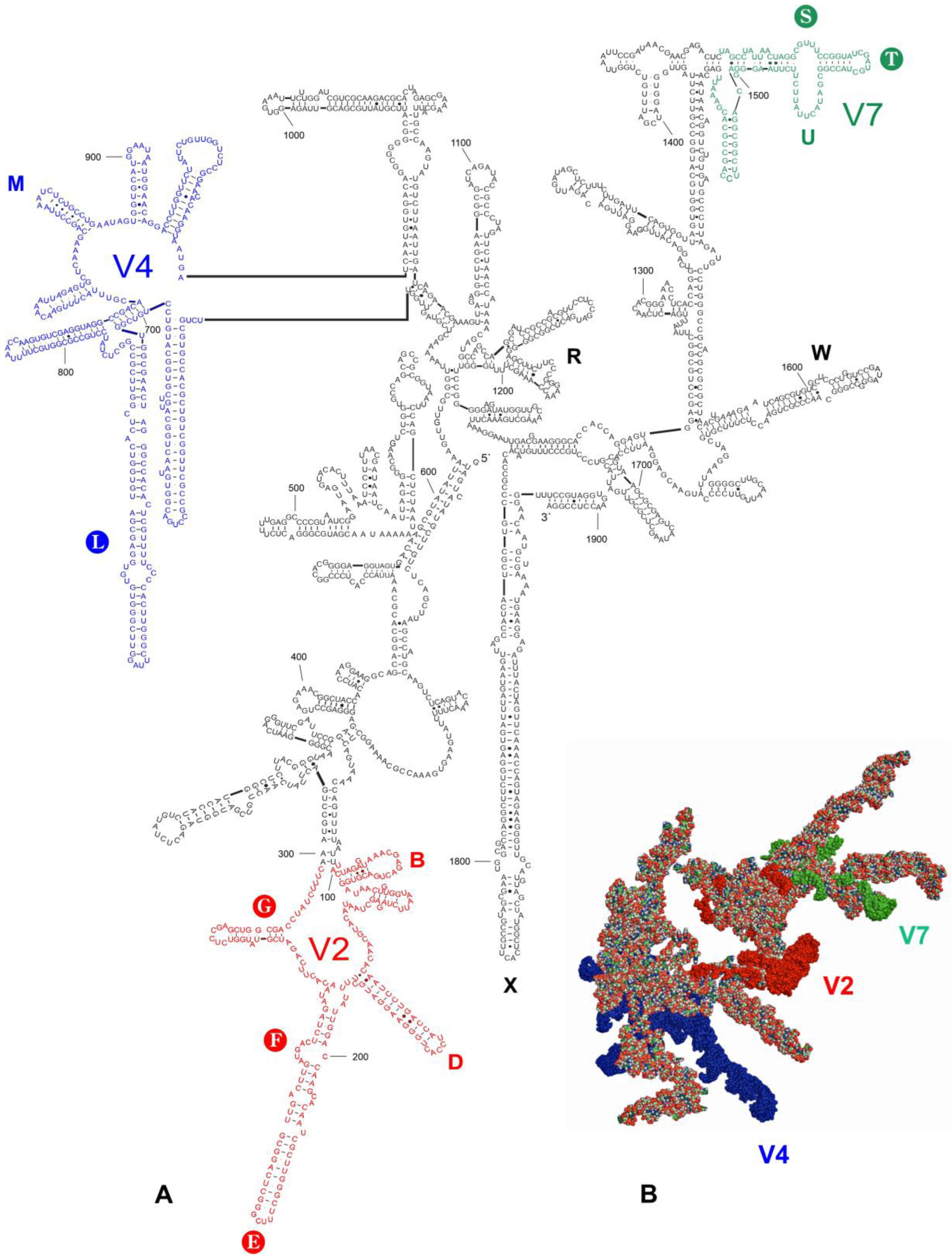
3.2. Secondary Structure Models
3.3. Tertiary Structure Models
4. Discussion
4.1. 18S rRNA Secondary and Tertiary Structures of Tingidae
4.2. Potential Apomorphies in Secondary and Tertiary Structures of LVRs
4.3. Identity and Systematic Position of the Tribe Acaltyptaini within the Subfamily Tinginae
5. Conclusions
- The results of the present molecular analyses (phylogenetic and structural) validated the recognition of the tribe Acalyptaini within the subfamily Tinginae.
- The monophyly of the subfamily Cantacaderinae and its basal position within the family Tingidae were indicated, as well as the position of the tribe Litadeini as sister to all other Tinginae.
- The structural analysis of the predicted tertiary structures of the entire 18S rRNA confirmed the proposed hypothesis that the in vivo configuration of this gene is likely not predictable using only secondary structure models and existing software. However, this may result from small nucleolar RNAs (snoRNAs) activities that can affect changes in the tertiary structures of ribosomal genes.
- The results showed that two LVRs (LVR X and LVR L) of the hypervariable region V4 exhibited significant variability in the number of nucleotides and could be considered for apomorphic recognition.
- LVR L appeared to be the most appropriate for phylogenetic relationship analysis within the family Tingidae when considering the secondary and tertiary structure models suitable for identifying morpho-molecular apomorphies.
- The subfamily Cantacaderinae exhibited the highest number of morpho-molecular autapomorphies in the secondary and tertiary structures of the 18S rRNA. In particular, the absence of the entire subregion LB in this subfamily is the first example of such an extensive deletion in the 18S rDNA sequence in Heteroptera.
- The tertiary structure of the 18S rRNA exhibited evolutionary properties, which were not detectable in the primary or secondary structures. Therefore, the results of rRNA tertiary structure analyses for phylogenetic considerations are promising. Therefore, including the methods of rRNA tertiary structure analyses in the phylogenetic evaluations in other groups of Heteroptera is strongly suggested.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuh, R.T.; Weirauch, C.H. True Bugs of the World (Hemiptera: Heteroptera): Classification and Natural History, 2nd ed.; Monograph Series; Siri Scientific Press: Rochdale, UK, 2020; Volume 8, 767p. [Google Scholar]
- Schuh, R.T.; Cassis, G.; Guilbert, E. Description of the first recent macropterous species of Vianaidinae (Heteroptera: Tingidae) with comments on the phylogenetic relationships of the family within the Cimicomorpha. J. N. Y. Entomol. Soc. 2006, 114, 38–53. [Google Scholar] [CrossRef]
- Lis, B. Phylogeny and classification of Cantacaderini [=Cantacaderidae stat. nov.] (Hemiptera: Tingoidea). Ann. Zool. 1999, 49, 157–196. [Google Scholar]
- Guilbert, E. Phylogeny of Cantacaderinae (Heteroptera: Tingidae) revisited after the description of a new genus and new species from New Caledonia. Eur. J. Entomol. 2012, 109, 111–116. [Google Scholar] [CrossRef]
- Drake, C.J.; Ruhoff, F.A. Lacebugs of the World: A Catalog (Hemiptera: Tingidae). USA Natl. Mus. Bull. 1965, 243, 1–634. [Google Scholar] [CrossRef]
- Froeschner, R.C. Zoogeographic and systematic notes on the lace bug tribe Litadeini, with the description of the new genus Stragulotingis (Hemiptera: Tingidae). Great Basin Nat. 1969, 29, 129–132. [Google Scholar]
- Froeschner, R.C. Lace Bug Genera of the World, II: Subfamily Tinginae: Tribes Litadeini and Ypsotingini (Heteroptera; Tingidae). Smithson. Contrib. Zool. 2001, 611, 1–28. [Google Scholar] [CrossRef]
- Guilbert, E.; Damgaard, J.; D’Haese, C.A. Phylogeny of the lacebugs (Insecta: Heteroptera: Tingidae) using morphological and molecular data. Syst. Entomol. 2014, 39, 431–441. [Google Scholar] [CrossRef]
- Golub, V.B.; Golub, N.V. On the status of the genera complex Acalypta, Dictyonota, Kalama and Derephysia (Heteroptera: Tingidae: Tinginae) having common morphological and karyological features. Zoosyst. Ross. 2019, 28, 228–237. [Google Scholar] [CrossRef]
- Gapon, D.A.; Golub, V.B.; Knudson, A.H. Case 3813—Acalyptini Thomson, 1859 (Hexapoda, Coleoptera) and Acalyptini Blatchley, 1926 (Hexapoda, Heteroptera): Proposed removal of homonymy by emendation of the latter name to Acalyptaini. Bull. Zool. Nomencl. 2019, 76, 175–178. [Google Scholar] [CrossRef]
- Lis, B.; Zielińska, A.; Lis, J.A. The King’s Lace Bug Recaredus rex Distant, 1909 (Hemiptera: Heteroptera: Tingidae): Systematic Position, First Palaearctic and Afrotropical Records, and Ecological Niche Modelling. Insects 2022, 13, 558. [Google Scholar] [CrossRef]
- Golub, N.V.; Golub, V.B.; Anokhin, B.A.; Kuznetsova, V.G. Comparative Cytogenetics of Lace Bugs (Tingidae, Heteroptera): New Data and a Brief Overview. Insects 2022, 13, 608. [Google Scholar] [CrossRef] [PubMed]
- Lis, B.; Parveen, S.; Ramamurthy, V.V. Redescription of the Oriental lace-bug Recaredus rex Distant, 1909 (Hemiptera: Tingidae: Tinginae), and its new tribal assignment, with a key to Ypsotingini. Zootaxa 2013, 3702, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Lis, B. Comparative studies on the ductus seminis of aedeagus in Tingoidea (Hemiptera: Heteroptera). Pol. J. Entomol. 2004, 73, 245–258. [Google Scholar]
- Lis, J.A. Molecular Apomorphies in the Secondary and Tertiary Structures of Length-Variable Regions (LVRs) of 18S rRNA Shed Light on the Systematic Position of the Family Thaumastellidae (Hemiptera: Heteroptera: Pentatomoidea). Int. J. Mol. Sci. 2023, 24, 7758. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, C.; Schuh, R.T.; Cassis, G.; Wheeler, W.C. Revisiting habitat and lifestyle transitions in Heteroptera (Insecta: Hemiptera): Insights from a combined morphological and molecular phylogeny. Cladistics 2019, 35, 67–105. [Google Scholar] [CrossRef]
- Lis, J.A.; Ziaja, D.; Lis, B.; Gradowska, P.A. Non-monophyly of the “cydnoid” complex within Pentatomoidea (Hemiptera: Heteroptera) revealed by Bayesian phylogenetic analysis of nuclear rDNA sequences. Arthropod Syst. Phylogeny 2017, 75, 481–496. [Google Scholar]
- Giribet, G.; Carranza, S.; Bagui, J.; Riutort, M.; Ribera, C. First Molecular Evidence for the Existence of a Tardigrada + Arthropoda Clade. Mol. Biol. Evol. 1996, 13, 76–84. [Google Scholar] [CrossRef]
- Whiting, M.F.; Carpernter, J.C.; Wheeler, Q.D.; Wheeler, W.C. The Strepsiptera problem: Phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst. Biol. 1997, 46, 1–68. [Google Scholar] [CrossRef]
- Campbell, B.C.; Steffen-Campbell, J.D.; Sorensen, J.T.; Gill, R.J. Paraphyly of Homoptera and Auchenorrhyncha inferred from 18S rDNA nucleotide sequences. Syst. Entomol. 1995, 20, 175–194. [Google Scholar] [CrossRef]
- Black, W.C., IV; Klompen, J.S.H.; Keirans, J.E. Phylogenetic relationships among tick subfamilies based on the SSU nuclear rDNA gene. Mol. Phylogenet. Evol. 1997, 7, 129–144. [Google Scholar] [CrossRef]
- Barker, S.C.; Whiting, M.; Johnson, K.P.; Murrell, A. Phylogeny of the lice (Insecta, Phthiraptera) inferred from small subunit rRNA. Zool. Scr. 2003, 32, 407–414. [Google Scholar] [CrossRef]
- Yu, S.; Wang, Y.; Rédei, D.; Xie, Q.; Bu, W. Secondary structure models of 18S and 28S rRNAs of the true bugs based on complete rDNA sequences of Eurydema maracandica Oshanin, 1871 (Heteroptera, Pentatomidae). ZooKeys 2013, 319, 363–377. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, 232–235. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Yu, S.S.; Wang, Y.H.; Wu, Y.H.; Li, X.R.; Men, X.Y.; Zhang, Y.W.; Rédei, D.; Xie, Q.; Bu, W.J. The evolutionary position of Lestoniidae revealed by molecular autapomorphies in the secondary structure of rRNA besides phylogenetic reconstruction (Insecta: Hemiptera: Heteroptera). Zool. J. Linn. Soc. 2016, 177, 750–763. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Wang, J.; Homoptera, J.; Huang, Y.; Xiao, Y. 3dRNA v2. 0: An Updated Web Server for RNA 3D Structure Prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef] [PubMed]
- Biesiada, M.; Purzycka, K.J.; Szachniuk, M.; Blazewicz, J.; Adamiak, R.W. Automated RNA 3D structure prediction with RNAComposer. In RNA Structure Determination. Methods in Molecular Biology; Turner, D., Mathews, D., Eds.; Humana Press: New York, NY, USA, 2016; Volume 1490, pp. 199–215. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL, Version 2.4.0; Schrödinger, LLC.: New York, NY, USA, 2020; Available online: http://www.pymol.org/pymol (accessed on 11 May 2023).
- Xie, Q.; Tian, X.; Qin, Y.; Bu, W. Phylogenetic comparison of local length plasticity of the small subunit of nuclear rDNAs among all Hexapoda orders and the impact of hyper-length-variation on alignment. Mol. Phylogenet. Evol. 2009, 50, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Ouvrard, D.; Campbell, B.C.; Bourgoin, T.; Chan, K.L. 18S rRNA Secondary Structure and Phylogenetic Position of Peloridiidae (Insecta, Hemiptera). Mol. Phylogenet. Evol. 2000, 16, 403–417. [Google Scholar] [CrossRef]
- Xie, Q.; Tian, Y.; Zheng, L.; Bu, W. 18S rRNA hyper-elongation and the phylogeny of Euhemiptera (Insecta: Hemiptera). Mol. Phylogenet. Evol. 2008, 47, 463–471. [Google Scholar] [CrossRef]
- Kiss, T. Small Nucleolar RNAs An Abundant Group of Noncoding RNAs with Diverse Cellular Functions. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef]
- Zemann, A.; op de Bekke, A.; Kiefmann, M.; Brosius, J.; Schmitz, J. Evolution of small nucleolar RNAs in nematodes. Nucleic Acids Res. 2006, 34, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Zemann, A.; Churakov, G.; Kuhl, H.; Grützner, F.; Reinhardt, R.; Brosius, J. Retroposed SNOfall-A mammalian-wide comparison of platypus snoRNAs. Genome Res. 2008, 18, 1005–1010. [Google Scholar] [CrossRef]
- Bratkovič, T.; Božič, J.; Rogelj, B. Functional diversity of small nucleolar RNAs. Nucleic Acids Res. 2020, 48, 1627–1651. [Google Scholar] [CrossRef] [PubMed]
- Soulé, S.; Mellottee, L.; Arab, A.; Chen, C.; Martin, J.-R. Jouvence, a new small nucleolar RNA required in the gut extends lifespan in Drosophila. Nat. Commun. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.T.; Štys, P. Phylogenetic analysis of cimicomorphan family relationships (Heteroptera). J. N. Y. Entomol. Soc. 1991, 99, 298–350. [Google Scholar]
- Schuh, R.T.; Weirauch, C.; Wheeler, W.C. Phylogenetic relationships within the Cimicomorpha (Hemiptera: Heteroptera): A total-evidence analysis. Syst. Entomol. 2009, 34, 15–48. [Google Scholar] [CrossRef]
- Wang, Y.H.; Cui, Y.; Rédei, D.; Baňař, P.; Xie, Q.; Štys, P.; Damgaard, J.; Chen, P.P.; Yi, W.B.; Wang, Y.; et al. Phylogenetic divergences of the true bugs (Insecta: Hemiptera:Heteroptera), with emphasis on the aquatic lineages: The last piece of the aquatic insect jigsaw originated in the Late Permian/EarlyTriassic. Cladistics 2016, 32, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Steffen-Campbell, J.D.; Gill, R.J. Evolutionary origin of whiteflies (Hemiptera: Sternorrhyncha: Aleyrodidae) inferred from 18S rDNA sequences. Insect Mol. Biol. 1994, 3, 73–88. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon Group | Consensus Species | Number of Nucleotides | ||
|---|---|---|---|---|
| V2 | V4 | V7 | ||
| Outgroup | Adelphocoris lineolatus (Goeze, 1778) | 198 | 317 | 91 |
| Tinginae: Acalyptaini | Acalypta sauteri (Drake, 1942) | 200 | 321 | 90 |
| Tinginae: Litadeini | Nobarnus signatus (Distant, 1920) | 202 | 323 | 90 |
| Tinginae: Tingini | Tingis matsumurai (Takeya, 1962) | 200 | 320 | 90 |
| Cantacaderinae | Cantacader lethierryi (Scott, 1874) | 199 | 299 | 90 |
| Taxon Group | Number of Nucleotides | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B | D | E | F | G | L | M | S | T | U | R | W | X | |
| outgroup | 12 | 6 | 5 | 4 | 0 | 60–78 | 4 | 6 | 7 | 13 | 5 | 3 | 6 |
| Tinginae: Acalyptaini | 11 | 5 | 4 | 5 | 0 | 79 | 4 | 5 | 7 | 13 | 5 | 3 | 3–4 |
| Tinginae: Litadeini | 11 | 4 | 5 | 5 | 0 | 81 | 4 | 5 | 7 | 13 | 5 | 3 | 6 |
| Tinginae: Tingini | 11 | 5 | 4–5 | 5 | 0 | 63–79 | 4 | 5 | 7 | 13 | 5 | 3 | 5–6 |
| Cantacaderinae | 11 | 5 | 5 | 4 | 0 | 57 | 4 | 5 | 7 | 13 | 5 | 3 | 5 |
| Taxon Group | Consensus Species | Total Length | Number of Nucleotides of the LVR L Subregions | |||||
|---|---|---|---|---|---|---|---|---|
| L2 | LA (A1 + A2) | LB (B1 + B2) | LC (C1 + C2) | LD (D1 + D2) | LE (E1 + E2) | |||
| outgroup | Adelphocoris lineolatus | 74 | 4 | 18 (9 + 9) | 19 (10 + 9) | 7 (3 + 4) | 11 (6 + 5) | 15 (8 +7) |
| Acalyptaini | Acalypta sauteri | 79 | 4 | 19 (10 + 9) | 21 (11+ 10) | 8 (4 + 4) | 10 (5 + 5) | 16 (8 + 8) |
| Litadeini | Nobarnus signatus | 81 | 4 | 19 (10 + 9) | 24 (13 + 11) | 8 (4 + 4) | 10 (5 + 5) | 16 (8 + 8) |
| Tingini | Tingis matsumurai | 78 | 5 | 19 (10 + 9) | 22 (11 + 11) | 8 (3 + 5) | 10 (6 + 4) | 14 (7 + 7) |
| Cantacaderinae | Cantacader lethierryi | 57 | 4 | 18 (9 + 9) | 0 | 12 (7 + 5) | 14 (7 + 7) | 9 (4 + 5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lis, B.; Domagała, P.J.; Lis, J.A. Tribe Acalyptaini (Hemiptera: Tingidae: Tinginae) Revisited: Can Apomorphies in Secondary and Tertiary Structures of 18S rRNA Length-Variable Regions (LVRs) Support Tribe Validity? Insects 2023, 14, 600. https://doi.org/10.3390/insects14070600
Lis B, Domagała PJ, Lis JA. Tribe Acalyptaini (Hemiptera: Tingidae: Tinginae) Revisited: Can Apomorphies in Secondary and Tertiary Structures of 18S rRNA Length-Variable Regions (LVRs) Support Tribe Validity? Insects. 2023; 14(7):600. https://doi.org/10.3390/insects14070600
Chicago/Turabian StyleLis, Barbara, Paweł J. Domagała, and Jerzy A. Lis. 2023. "Tribe Acalyptaini (Hemiptera: Tingidae: Tinginae) Revisited: Can Apomorphies in Secondary and Tertiary Structures of 18S rRNA Length-Variable Regions (LVRs) Support Tribe Validity?" Insects 14, no. 7: 600. https://doi.org/10.3390/insects14070600
APA StyleLis, B., Domagała, P. J., & Lis, J. A. (2023). Tribe Acalyptaini (Hemiptera: Tingidae: Tinginae) Revisited: Can Apomorphies in Secondary and Tertiary Structures of 18S rRNA Length-Variable Regions (LVRs) Support Tribe Validity? Insects, 14(7), 600. https://doi.org/10.3390/insects14070600