Comparative Identification of MicroRNAs in Apis cerana cerana Workers’ Midguts in Response to Nosema ceranae Invasion



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of N. ceranae Spores
2.2. Experimental Inoculation of Honeybees
2.3. Small RNA Isolation, cDNA Library Construction and Deep Sequencing
2.4. Quality Control and Sequencing Data Analysis
2.5. Analysis of DEmiRNAs
2.6. Prediction of the Target mRNAs of DEmiRNAs and Construction of miRNA-mRNA Regulation Networks
2.7. GO and KEGG Pathway Analyses of DEmiRNA Target mRNAs
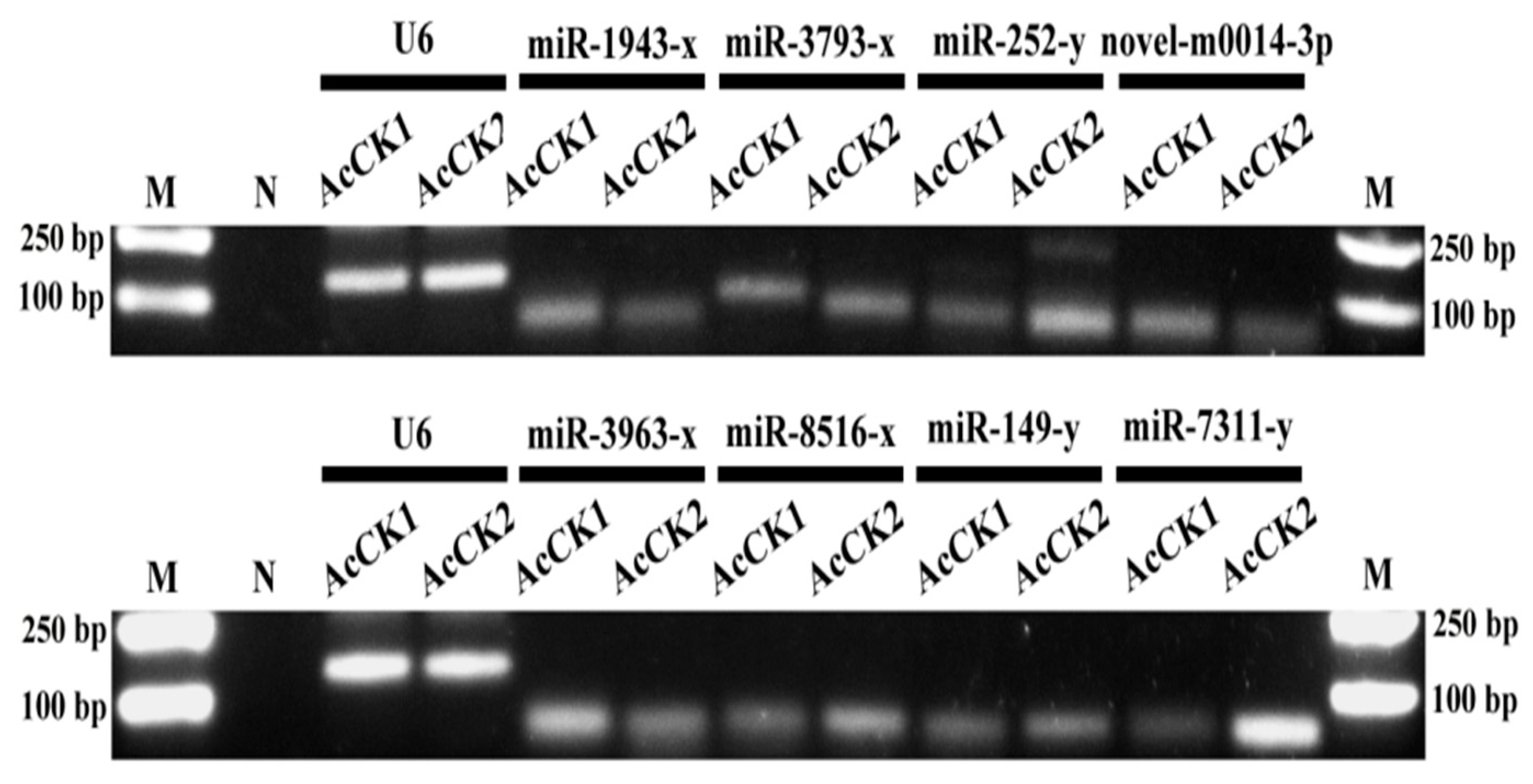
2.8. Stem-Loop RT-PCR Confirmation of miRNAs
2.9. RT-qPCR Validation of DEmiRNAs
2.10. Statistical Data Analysis
3. Results
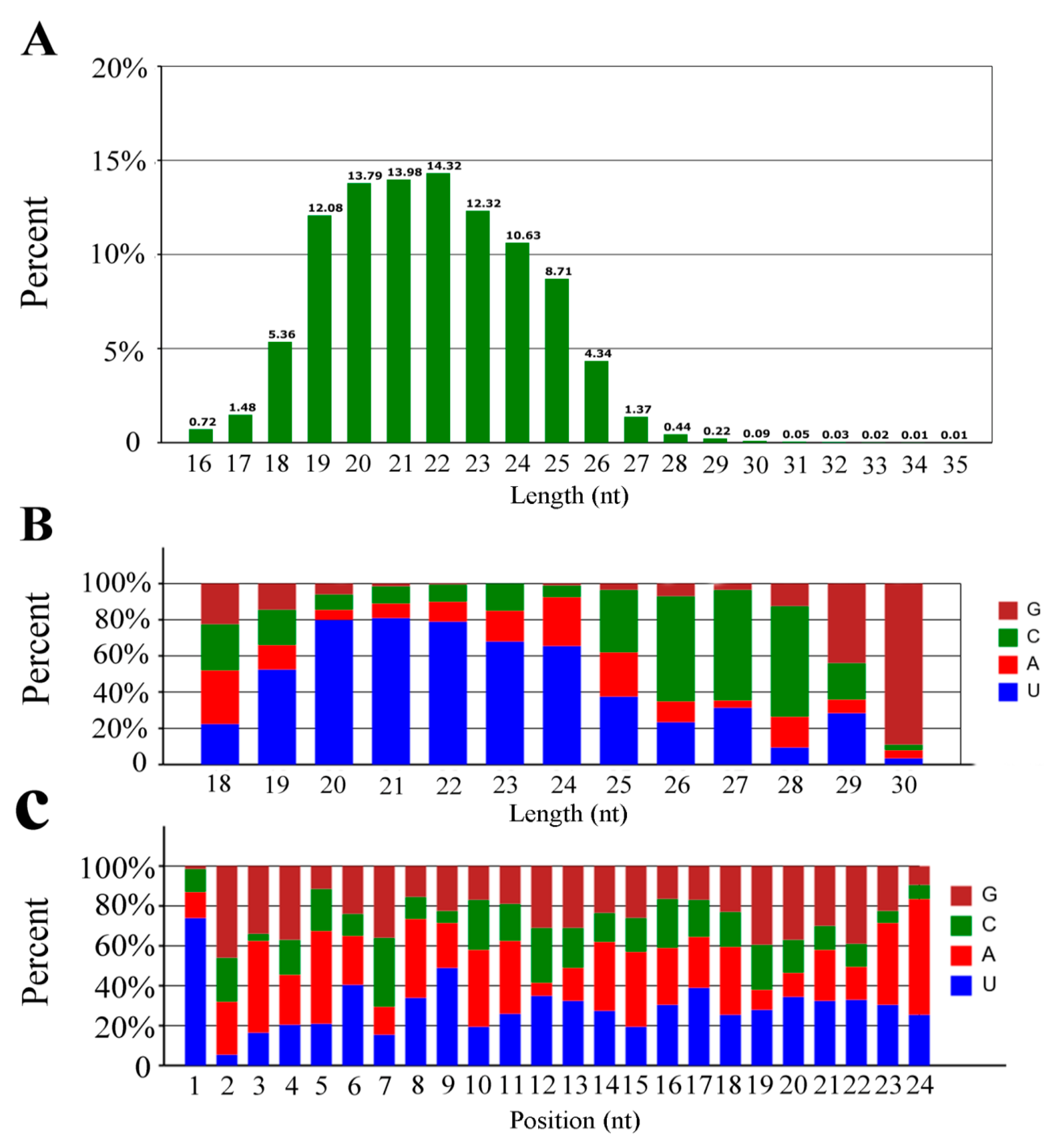
3.1. Overview of the Small RNA Library from A. c. cerana Workers’ Guts
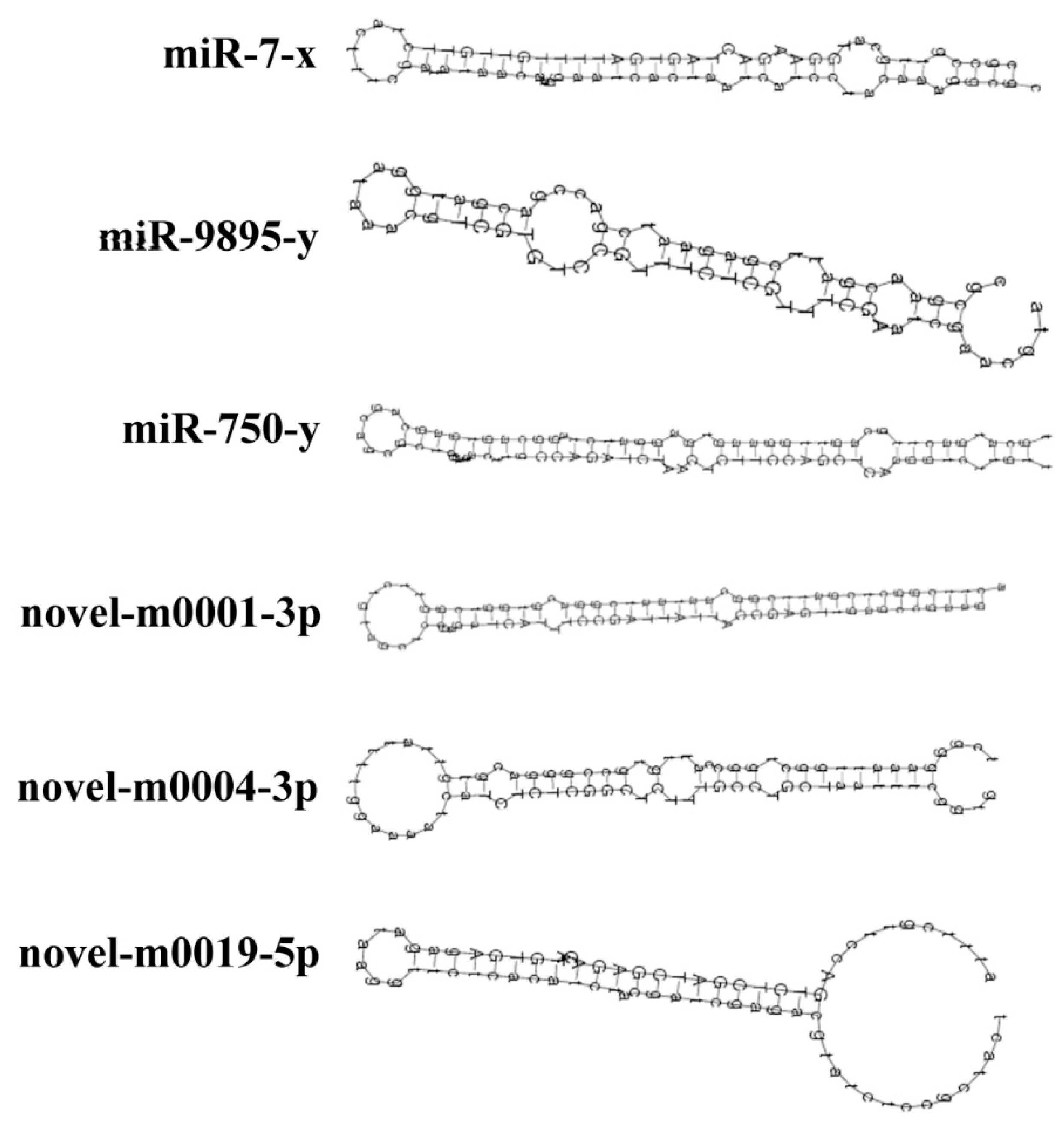
3.2. Identification of Known and Novel miRNAs in A. c. cerana Worker’s Midgut
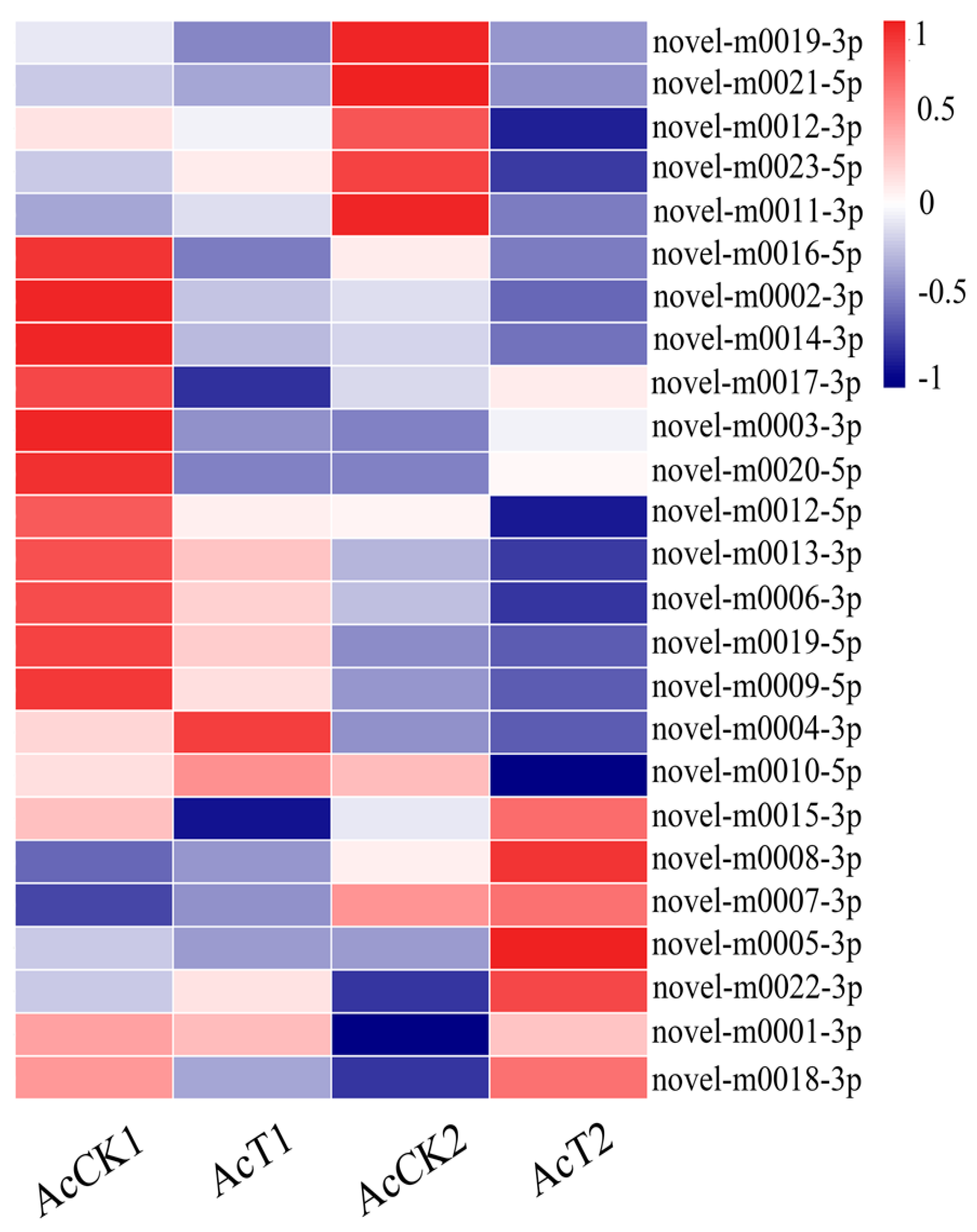
3.3. Differentially Expressed A. c. cerana miRNAs Induced by N. ceranae Invasion
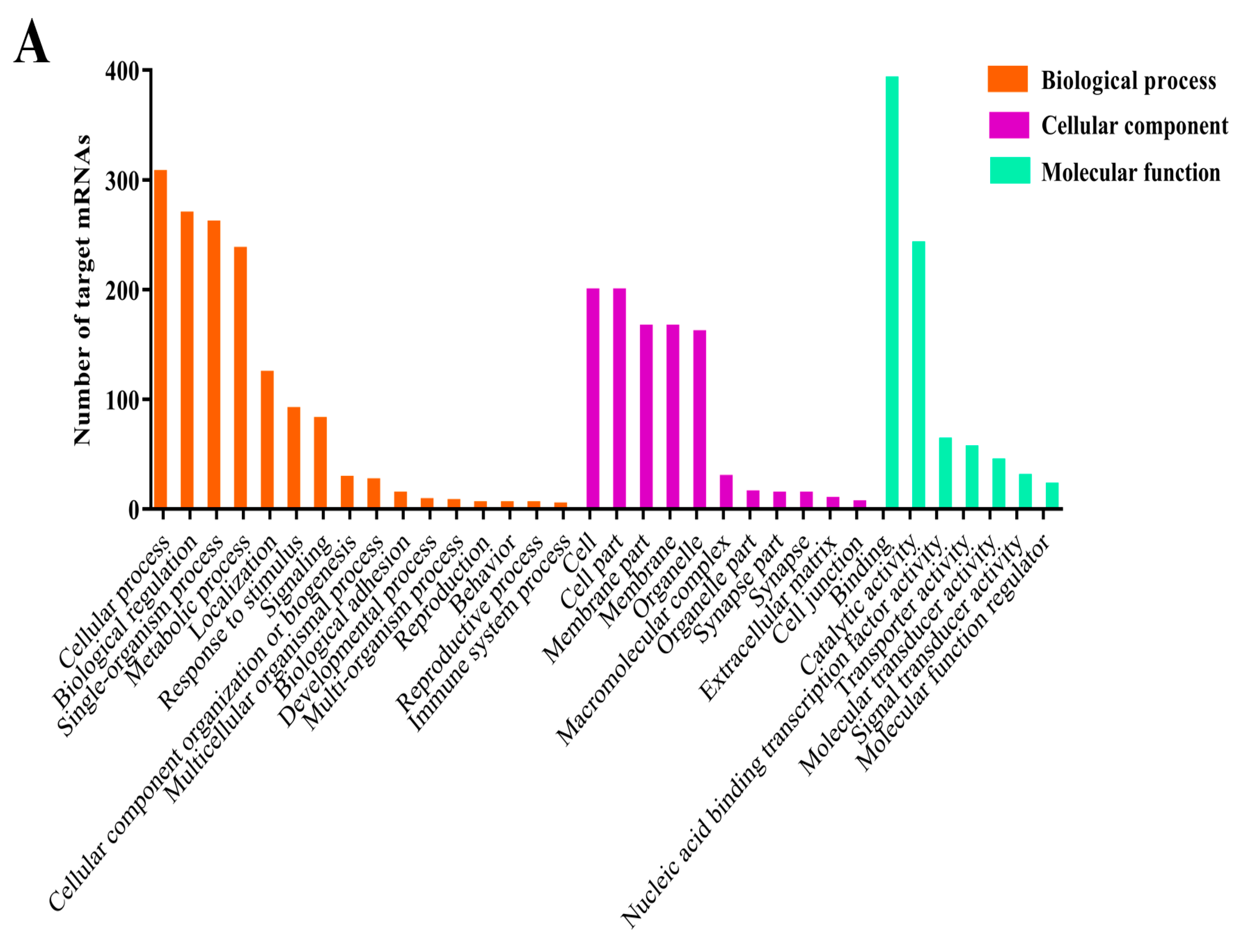
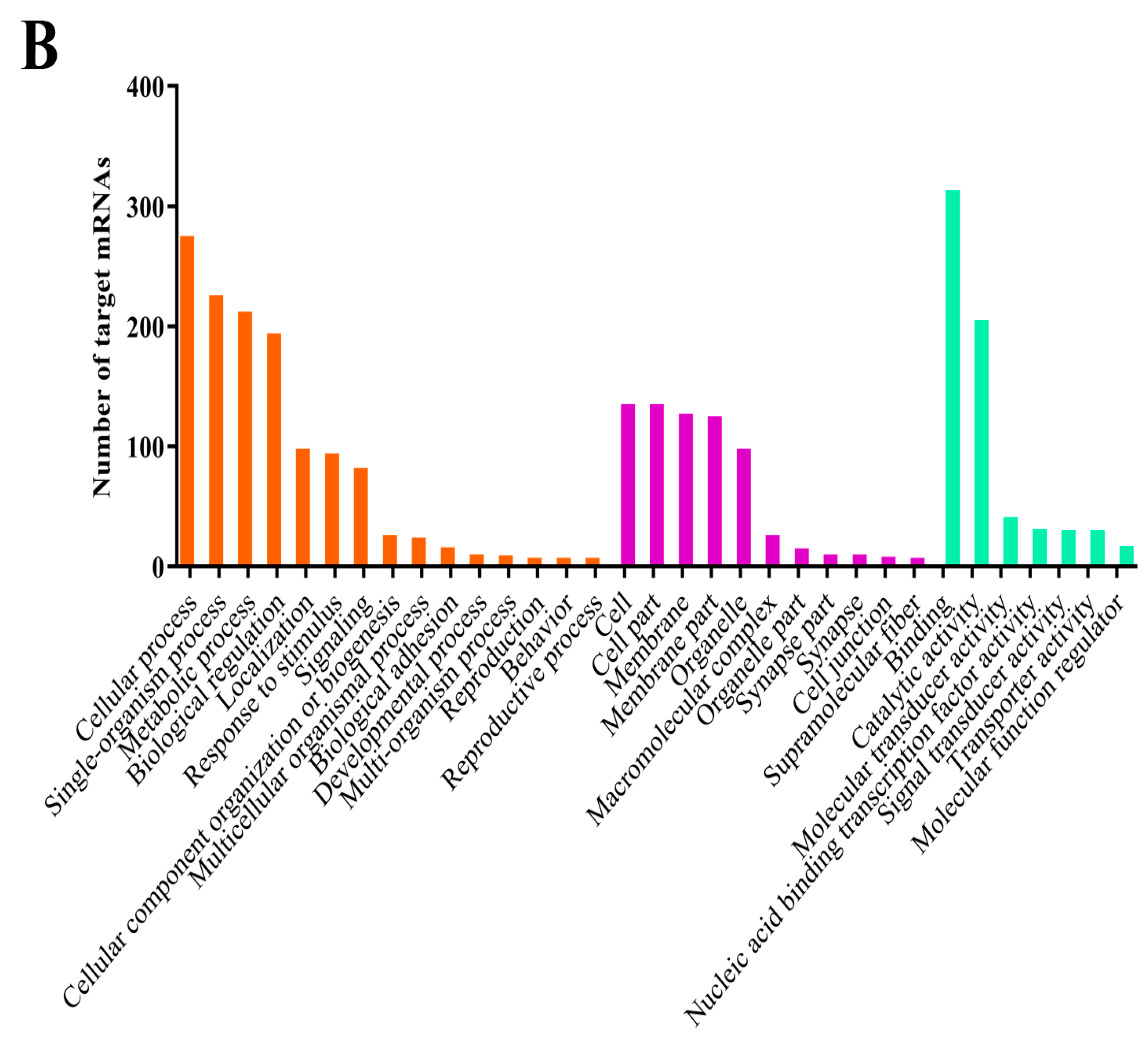
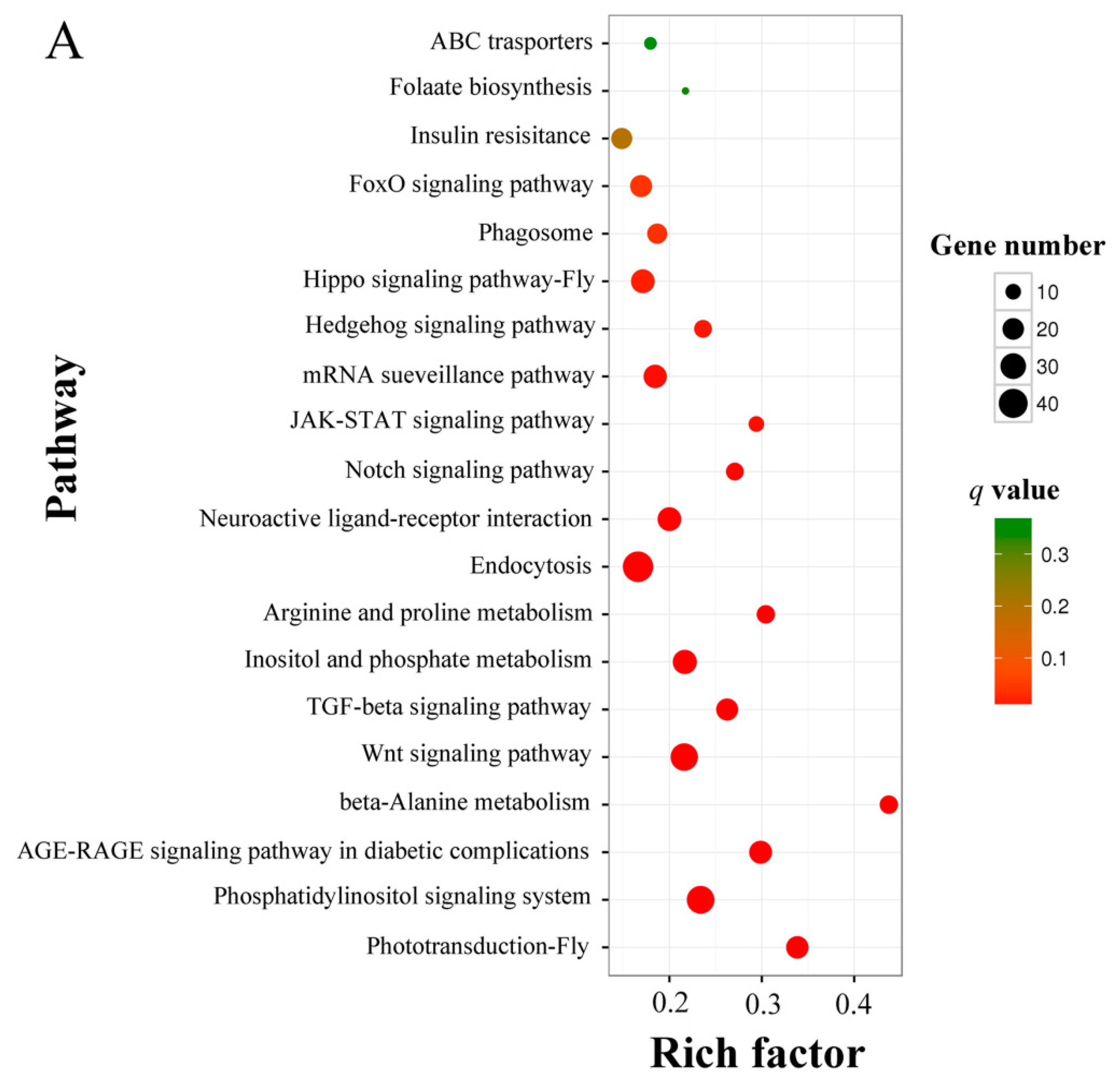
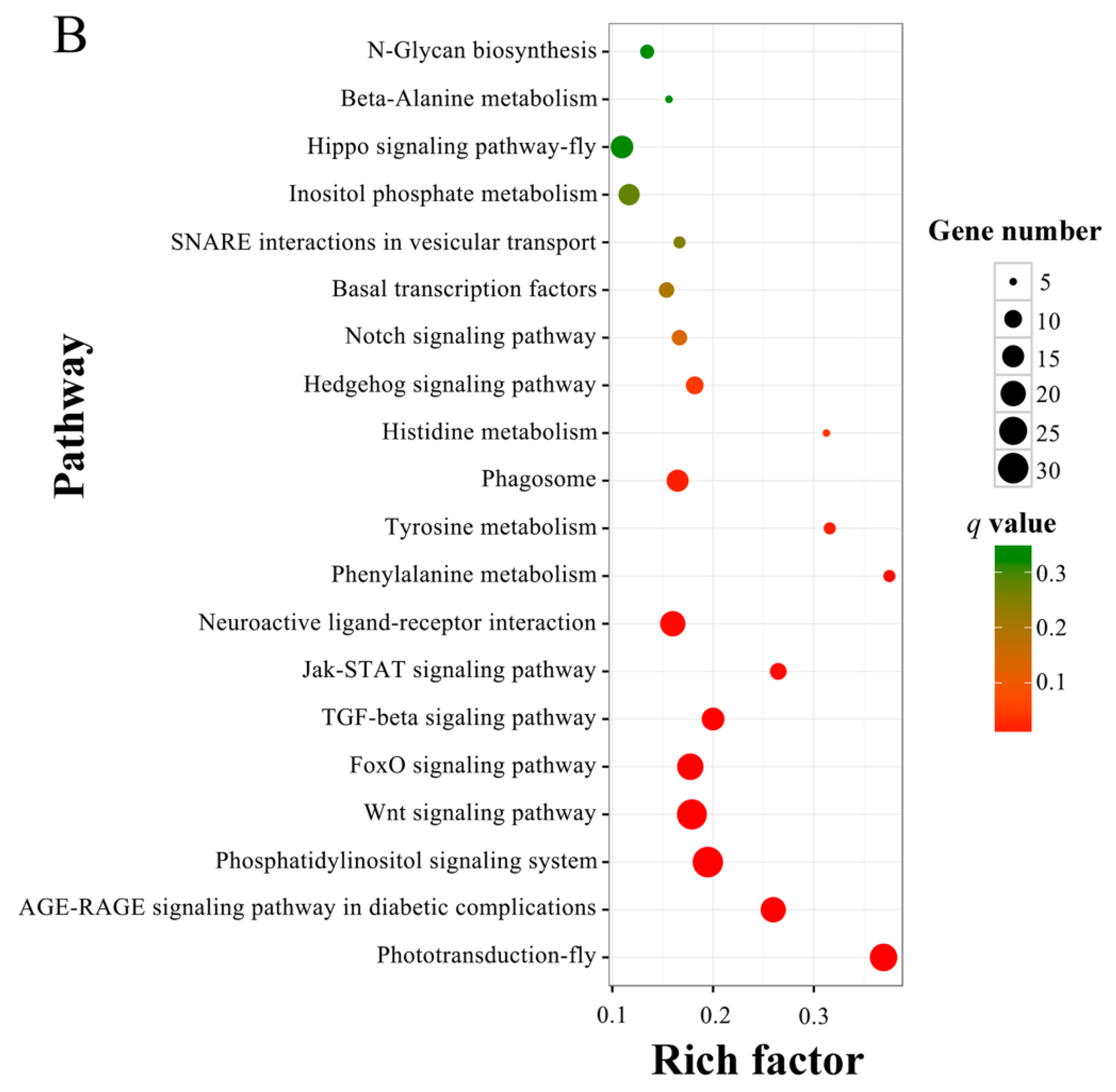
3.4. Prediction and Enrichment Analysis of the Target mRNAs of A. c. cerana DEmiRNAs
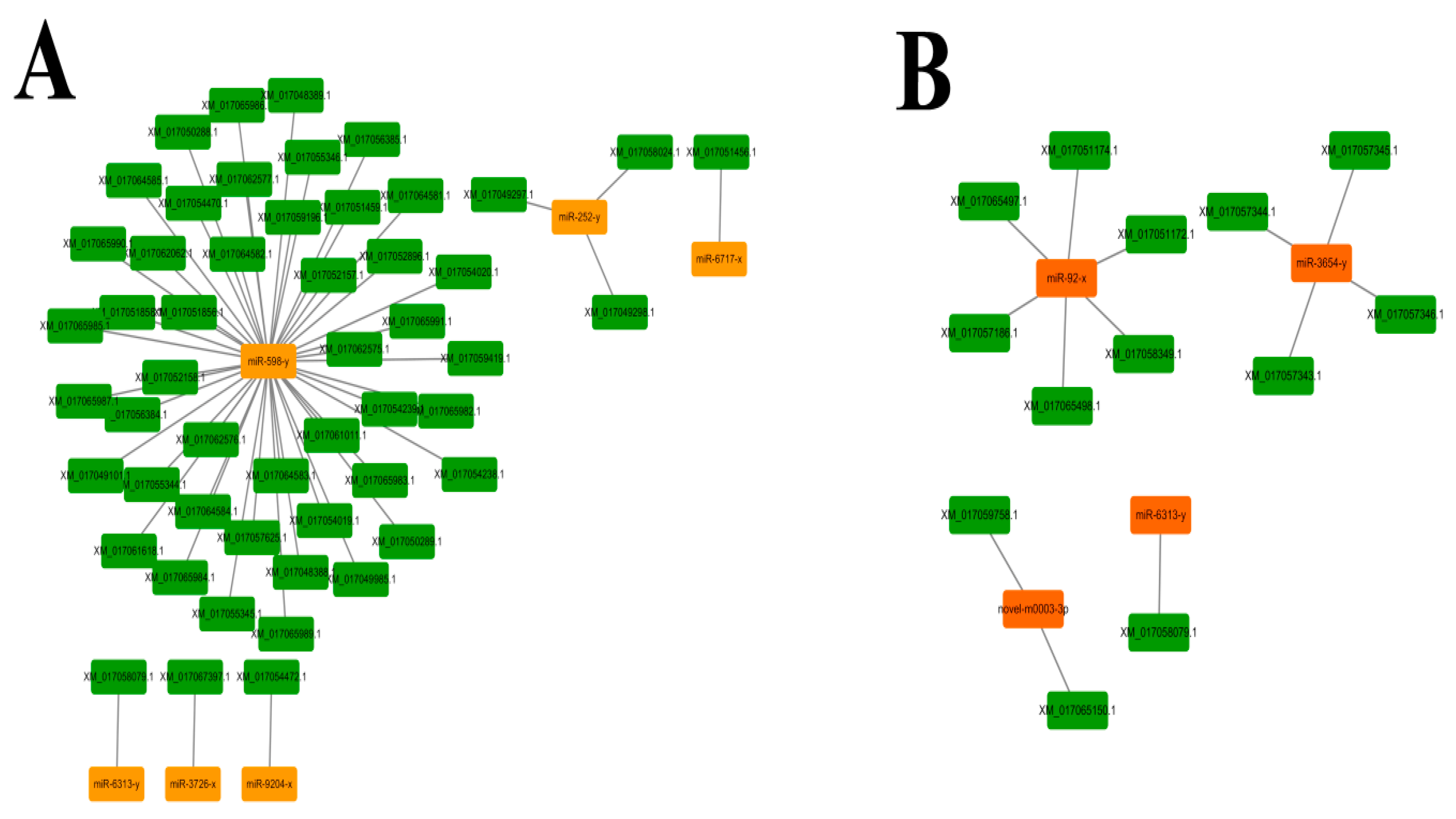
3.5. Regulatory Networks between A. c. cerana DEmiRNAs and Their Target mRNAs
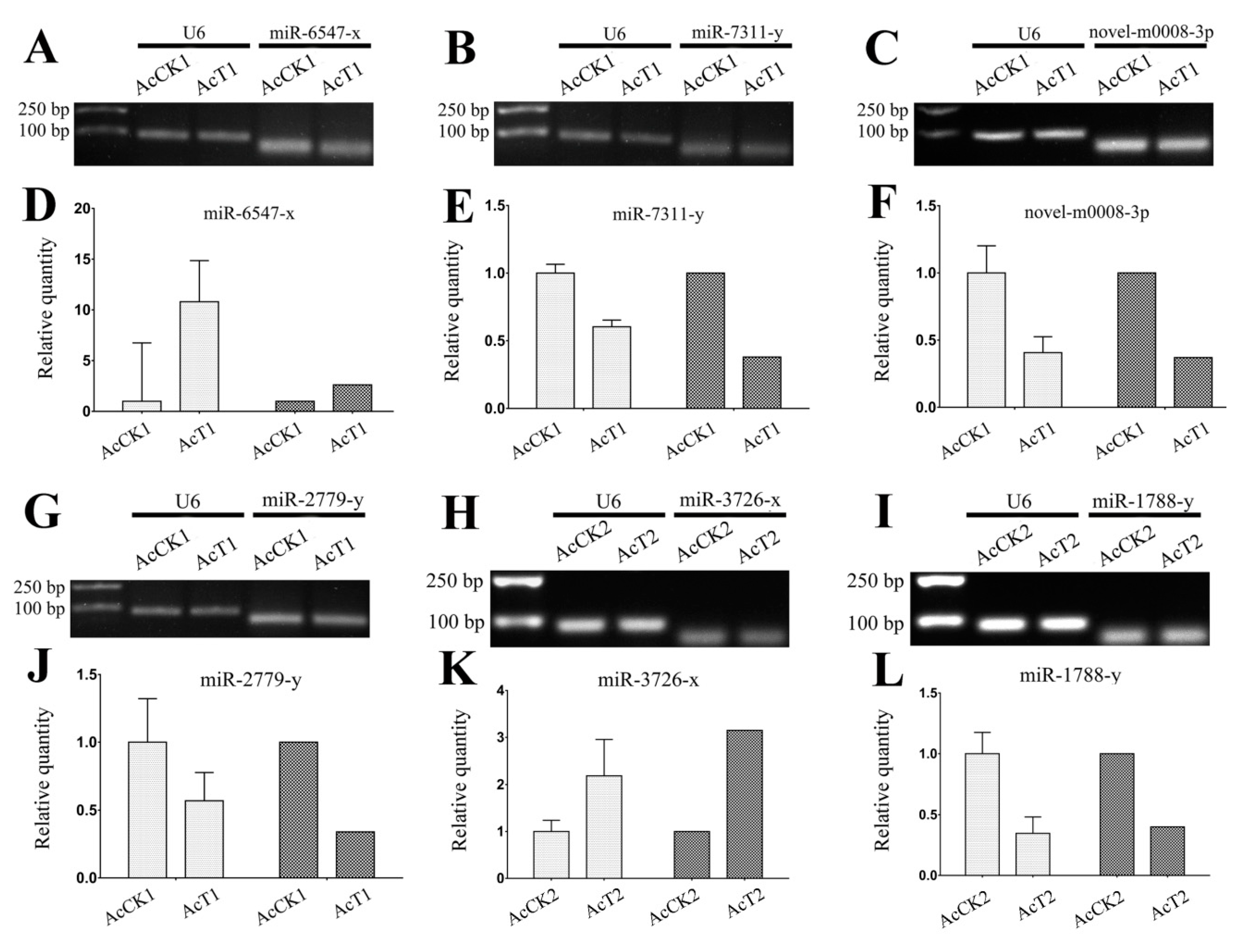
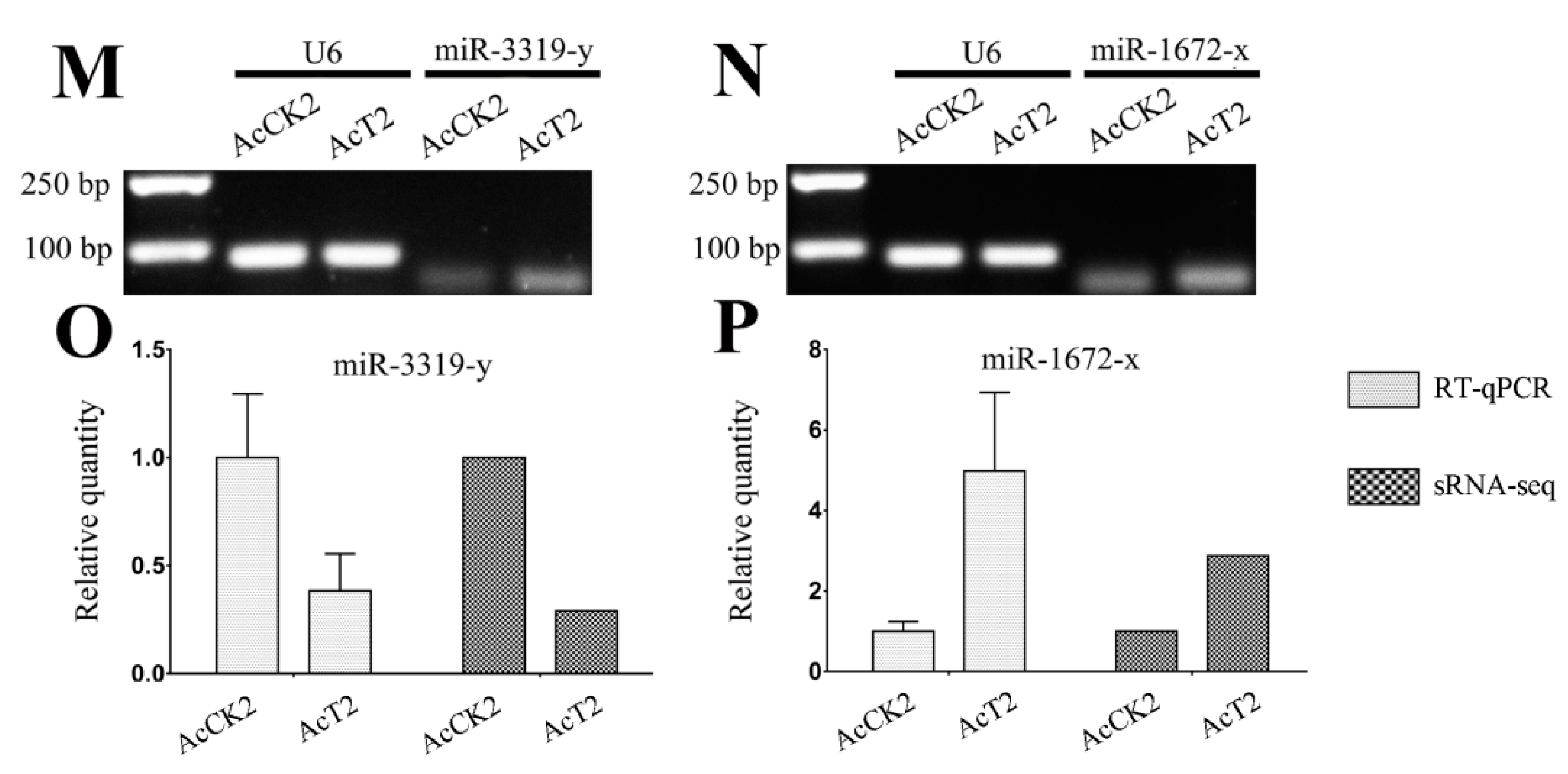
3.6. Verification of A. c. cerana DEmiRNAs via RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bromenshenk, J.J.; Henderson, C.B.; Seccomb, R.A.; Welch, P.M.; Debnam, S.E.; Firth, D.R. Bees as biosensors: Chemosensory ability, honey bee monitoring systems, and emergent sensor technologies derived from the pollinator syndrome. Biosensors 2015, 5, 678–711. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ke, L.; Wang, Z.; Zeng, Z. Dynamic transcriptome landscape of Asian domestic honeybee (Apis cerana) embryonic development revealed by high-quality RNA sequencing. BMC Dev. Biol. 2018, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Honey Bee Genome Sequencing Consortium. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genomics 2014, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Jung, J.W.; Choi, B.S.; Jayakodi, M.; Lee, J.; Lim, J.; Yu, Y.; Choi, Y.S.; Lee, M.L.; Park, Y.; et al. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics 2014, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, B.P.; Wongsiri, S. Asian Honey Bees: Biology, Conservation, and Human Interactions; Harvard University Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Guo, R.; Chen, D.; Diao, Q.; Xiong, C.; Zheng, Y.; Hou, C. Transcriptomic investigation of immune responses of the Apis cerana cerana larval gut infected by Ascosphaera apis. J. Invertebr. Pathol. 2019, 166, 107210. [Google Scholar] [CrossRef] [PubMed]
- Wittner, M.; Weiss, L.M. The Microsporidia and Microsporidiosis; ASM Press: Washington, DC, USA, 1999. [Google Scholar]
- Visvesvara, G.S. In vitro cultivation of microsporidia of clinical importance. Clin. Microbiol. Rev. 2002, 15, 401–413. [Google Scholar] [CrossRef]
- Chen, Y.P.; Evans, J.D.; Murphy, C.; Gutell, R.; Zuker, M.; Gundensen-Rindal, D.; Pettis, J.S. Morphological, molecular, and phylogenetic characterization of Nosema ceranae, a microsporidian parasite isolated from the European honey bee, Apis mellifera. J. Eukaryot. Microbiol. 2010, 56, 142–147. [Google Scholar] [CrossRef]
- Adl, S.M.; Simpson, A.G.B.; Farmer, M.A.; Andersen, R.A.; Anderson, O.R.; Barta, J.R.; Bowser, S.S.; Brugerolle, G.; Fensome, R.A.; Fredericq, S.; et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot. Microbiol. 2005, 52, 399–451. [Google Scholar] [CrossRef]
- Fries, I.; Feng, F.; da Silva, A.; Slemenda, S.B.; Pieniazek, N.J. Nosema ceranae nsp (Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (Hymenoptera, Apidae). Eur. J. Protistol. 1996, 32, 356–365. [Google Scholar] [CrossRef]
- Higes, M.; Martin-Hernandez, R.; Meana, A. Nosema ceranae, a new microsporidian parasite in honeybees in Europe. J. Invertebr. Pathol. 2006, 92, 93–95. [Google Scholar] [CrossRef]
- Huang, W.F.; Jiang, J.H.; Chen, Y.W.; Wang, C.H. A Nosema ceranae isolate from the honeybee Apis mellifera. Apidologie 2007, 38, 30–37. [Google Scholar] [CrossRef]
- Klee, J.; Besana, A.M.; Genersch, E.; Gisder, S.; Nanetti, A.; Tam, D.Q.; Chinh, T.X.; Puerta, F.; Ruz, J.M.; Kryger, P.; et al. Widespread dispersal of the microsporidian Nosema ceranae, an emergent pathogen of the western honey bee, Apis mellifera. J. Invertebr. Pathol. 2007, 96, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Evans, J.D.; Smith, I.B.; Pettis, J.S. Nosema ceranae is a long-present and wide-spread microsporidian infection of the European honey bee (Apis mellifera) in the United States. J. Invertebr. Pathol. 2008, 97, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Alaux, C.; Brunet, J.L.; Dussaubat, C.; Mondet, F.; Tchamitchan, S.; Cousin, M.; Brillard, J.; Baldy, A.; Belzunces, L.P.; Le Conte, Y. Interactions between Nosema microspores and a neonicotinoid weaken honeybees (Apis mellifera). Environ. Microbiol. 2010, 12, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Pettis, J.S.; Vanengelsdorp, D.; Johnson, J.; Dively, G. Pesticide exposure in honey bees results in increased levels of the gut pathogen Nosema. Naturwissenschaften 2012, 99, 153–158. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef]
- Brummer, A.; Hausser, J. MicroRNA binding sites in the coding region of mRNAs: Extending the repertoire of post-transcriptional gene regulation. Bioessays 2014, 36, 617–626. [Google Scholar] [CrossRef]
- Li, G.; Wu, X.; Qian, W.; Cai, H.; Sun, X.; Zhang, W.; Tan, S.; Wu, Z.; Qian, P.; Ding, K.; et al. CCAR1 5′ UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res. 2016, 26, 655–673. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Yang, X.; Liu, Y.; Shi, Y.; Ren, J.; Guleng, B. MiRNA423-5p regulates cell proliferation and invasion by targeting trefoil factor 1 in gastric cancer cells. Cancer Lett. 2014, 347, 98–104. [Google Scholar] [CrossRef]
- Hammond, S.M. MicroRNAs as oncogenes. Curr. Opin. Genet. Dev. 2006, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Cristino, A.S.; Tanaka, E.D.; Rubio, M.; Piulachs, M.D.; Belles, X. Deep sequencing of organ- and stage-specific microRNAs in the evolutionarily basal insect Blattella germanica (L.) (Dictyoptera, Blattellidae). PLoS ONE 2011, 6, e19350. [Google Scholar] [CrossRef] [PubMed]
- Wagschal, A.; Najafishoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; deLemos, A.S.; Black, J.C.; Ramírez, C.M.; Li, Y.; Tewhey, R.; et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L.; Sharp, P.A. MicroRNA functions in stress responses. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Xu, S.; Liu, X.; Zhang, Q.; Xu, X.; Liu, M.; Hua, M.; Li, N.; Yao, H.; Cao, X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat. Immunol. 2011, 12, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, Y.; Rui, W.W.; Schwarz, R.S.; Evans, J.D. Honey bee microRNAs respond to infection by the microsporidian parasite Nosema ceranae. Sci. Rep. 2015, 5, 17494. [Google Scholar] [CrossRef] [PubMed]
- Higes, M.; Garcíapalencia, P.; Martínhernández, R.; Meana, A. Experimental infection of Apis mellifera honeybees with Nosema ceranae (Microsporidia). J. Invertebr. Pathol. 2007, 94, 211–217. [Google Scholar] [CrossRef]
- Cornman, R.S.; Chen, Y.P.; Schatz, M.C.; Street, C.; Zhao, Y.; Desany, B.; Egholm, M.; Hutchison, S.; Pettis, J.S.; Lipkin, W.I.; et al. Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog. 2009, 5, e1000466. [Google Scholar] [CrossRef]
- Fries, I.; Chauzat, M.P.; Chen, Y.P.; Doublet, V.; Genersch, E.; Gisder, S.; Higes, M.; McMahon, D.P.; Martín-Hernández, R.; Natsopoulou, M.; et al. Standard methods for Nosema research. J. Apic. Res. 2013, 52, 1–28. [Google Scholar] [CrossRef]
- Forsgren, E.; Fries, I. Comparative virulence of Nosema ceranae and Nosema apis in individual European honey bees. Vet. Parasitol. 2010, 170, 212–217. [Google Scholar] [CrossRef]
- Benjeddou, M.; Leat, N.; Allsopp, M.; Davison, S. Detection of acute bee paralysis virus and black queen cell virus from honeybees by reverse transcriptase PCR. Appl. Environ. Microbiol. 2001, 67, 2384–2387. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E. Development of a rapid and sensitive RT-PCR method for the detection of deformed wing virus, a pathogen of the honeybee (Apis mellifera). Vet. J. 2005, 169, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Ribière, M.; Triboulot, C.; Mathieu, L.; Aurières, C.; Faucon, J.P.; Pépin, M. Molecular diagnosis of chronic bee paralysis virus infection. Apidologie 2002, 33, 339–351. [Google Scholar] [CrossRef]
- Stoltz, D.; Shen, X.R.; Boggis, C.; Sisson, G. Molecular diagnosis of Kashmir bee virus infection. J. Apic. Res. 2015, 34, 153–160. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.L.; dePanmphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef]
- Hofacker, I.L. RNA secondary structure analysis using the Vienna RNA package. Curr. Protoc. Bioinform. 2009, 26, 12.2.1–12.2.16. [Google Scholar]
- Zhu, E.; Zhao, F.; Xu, G.; Hou, H.; Zhou, L.; Li, X.; Sun, Z.; Wu, J. MirTools: MicroRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Res. 2010, 38, W392–W397. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA. org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Chen, C.F.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Anaysis of relative gene expression data using realtime quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef]
- Shepotinovskaya, I.V.; Uhlenbeck, O.C. Catalytic diversity of extended hammerhead ribozymes. Biochemistry 2008, 47, 7034–7042. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Dezulian, T.; Palatnik, J.F.; Huson, D.; Weigel, D. Conservation and divergence of microRNA families in plants. Genome Biol. 2005, 6, 1–25. [Google Scholar] [CrossRef]
- Ai, L.; Xu, M.J.; Chen, M.X.; Zhang, Y.N.; Chen, S.H.; Guo, J.; Cai, Y.C.; Zhou, X.N.; Zhu, X.Q.; Chen, J.X. Characterization of microRNAs in Taenia saginata of zoonotic significance by Solexa deep sequencing and bioinformatics analysis. Parasitol. Res. 2012, 110, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Wang, G.; Xie, Z.; Zhang, C.; Wang, J. Identification and characterization of microRNA in the dairy goat (Capra hircus) mammary gland by Solexa deep-sequencing technology. Mol. Biol. Rep. 2012, 39, 9361–9371. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jia, Y.; Wang, P.; Yang, Q.; Du, Q.; Chang, Z. Identification and profiling of Cyprinus carpio microRNAs during ovary differentiation by deep sequencing. BMC Genomics 2017, 18, 333–349. [Google Scholar] [CrossRef]
- Chen, X.; Yu, X.; Cai, Y.; Zheng, H.; Yu, D.; Liu, G.; Zhou, Q.; Hu, S.; Hu, F. Next-generation small RNA sequencing for microRNAs profiling in the honey bee Apis mellifera. Insect Mol. Biol. 2010, 19, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Peng, W.; Li, Z.; Li, W.; Li, L.; Pan, J.; Zhang, S.; Miao, Y.; Chen, S.; Su, S. Next-generation small RNA sequencing for microRNAs profiling in Apis mellifera: Comparison between nurses and foragers. Insect Mol. Biol. 2012, 21, 297–303. [Google Scholar] [CrossRef]
- Zondag, L.; Dearden, P.K.; Wilson, M.J. Deep sequencing and expression of microRNAs from early honeybee (Apis mellifera) embryos reveals a role in regulating early embryonic patterning. BMC Evol. Biol. 2012, 12, 211–223. [Google Scholar] [CrossRef]
- Qin, Q.H.; Wang, Z.L.; Tian, L.Q.; Gan, H.Y.; Zhang, S.W.; Zeng, Z.J. The integrative analysis of microRNA and mRNA expression in Apis mellifera, following maze-based visual pattern learning. Insect Sci. 2014, 21, 619–636. [Google Scholar] [CrossRef]
- Ashby, R.; Forêt, S.; Searle, I.; Maleszka, R. MicroRNAs in honey bee caste determination. Sci. Rep. 2016, 6, 18794–18808. [Google Scholar] [CrossRef]
- Shi, Y.Y.; Wu, X.B.; Huang, Z.Y.; Wang, Z.L.; Yan, W.Y.; Zeng, Z.J. Epigenetic modification of gene expression in honey bees by heterospecific gland secretions. PLoS ONE 2012, 7, e43727. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Edward, M. Cloning, characterization, and expression of microRNAs from the Asian malaria mosquito, Anopheles stephensi. BMC Genomics 2008, 9, 244. [Google Scholar] [CrossRef]
- Padmanabhan, C.; Zhang, X.; Jin, H. Host small RNAs are big contributors to plant innate immunity. Curr. Opin. Plant Biol. 2009, 12, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.P.; Singh, J.; Nagaraju, J. A baculovirus-encoded microRNA (miRNA) suppresses its host miRNA biogenesis by regulating the exportin-5 cofactor Ran. J. Virol. 2012, 86, 7867–7879. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Evans, J.D. Identification of microRNA-like small RNAs from fungal parasite Nosema ceranae. J. Invertebr. Pathol. 2015, 133, 107–109. [Google Scholar] [CrossRef]
- Evans, J.D.; Huang, Q. Interactions among host-parasite microRNAs during Nosema ceranae proliferation in Apis mellifera. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Wojcicka, A.; Swierniak, M.; Kornasiewicz, O.; Gierlikowski, W.; Maciag, M.; Kolanowska, M.; Kotlarek, M.; Gornicka, B.; Koperski, L.; Niewinski, G.; et al. Next generation sequencing reveals microRNA isoforms in liver cirrhosis and hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2014, 53, 208–217. [Google Scholar] [CrossRef]
- Lu, J.; Shen, Y.; Wu, Q.; Kumar, S.; He, B.; Shi, S.; Carthew, R.W.; Wang, S.M.; Wu, C.I. The birth and death of microRNA genes in Drosophila. Nat. Genet. 2008, 40, 351–355. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, D.; Pflugfelder, G.O.; Shen, J. Dpp signaling inhibits proliferation in the Drosophila wing by Omb-dependent regional control of bantam. Development 2013, 140, 2917–2922. [Google Scholar] [CrossRef]
- Wu, Y.C.; Lee, K.S.; Song, Y.; Gehrke, S.; Lu, B. The bantam microRNA acts through Numb to exert cell growth control and feedback regulation of Notch in tumor-forming stem cells in the Drosophila brain. PLoS Genet. 2017, 13, e1006785. [Google Scholar] [CrossRef]
- Dong, L.; Li, J.; Huang, H.; Yin, M.X.; Xu, J.; Li, P.; Lu, Y.; Wu, W.; Yang, H.; Zhao, Y.; et al. Growth suppressor lingerer regulates bantam microRNA to restrict organ size. J. Mol. Cell Biol. 2015, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Catarina, B.P.; Fernando, C.; Florence, J. The retinal determination gene Dachshund restricts cell proliferation by limiting the activity of the Homothorax-Yorkie complex. Development 2015, 142, 1470–1479. [Google Scholar] [CrossRef]
- Boulan, L.; Martín, D.; Milán, M. Bantam miRNA promotes systemic growth by connecting insulin signaling and ecdysone production. Curr. Biol. 2013, 23, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Okamura, K.; Tyler, D.M.; Phillips, M.D.; Chung, W.J.; Lai, E.C. The evolution and functional diversification of animal microRNA genes. Cell Res. 2008, 18, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Yin, J.; Zhang, D.; Yin, J.; Xiang, Z.; Xia, Q. MicroRNAs show diverse and dynamic expression patterns in multiple tissues of Bombyx mori. BMC Genomics 2010, 11, 85–96. [Google Scholar] [CrossRef]
- Martinez, N.J.; Ow, M.C.; Reece-Hoyes, J.S.; Barrasa, M.I.; Ambros, V.R.; Walhout, A.J. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res. 2008, 18, 2005–2015. [Google Scholar] [CrossRef]
- Tan, Y.; Yamada-Mabuchi, M.; Arya, R.; St Pierre, S.; Tang, W.; Tosa, M.; Brachmann, C.; White, K. Coordinated expression of cell death genes regulates neuroblast apoptosis. Development 2011, 138, 2197–2206. [Google Scholar] [CrossRef]
- Ling, L.; Ge, X.; Li, Z.; Zeng, B.; Xu, J.; Chen, X.; Shang, P.; James, A.A.; Huang, Y.; Tan, A. MiR-2 family targets awd and fng to regulate wing morphogenesis in Bombyx mori. RNA Biol. 2015, 12, 742–748. [Google Scholar] [CrossRef]
- Boutla, A.; Delidakis, C.; Tabler, M. Developmental defects by antisense-mediated inactivation of microRNAs 2 and 13 in Drosophila and the identification of putative target genes. Nucleic Acids Res. 2003, 31, 4973–4980. [Google Scholar] [CrossRef]
- Kwon, C.; Han, Z.; Olson, E.N.; Srivastava, D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 18986–18991. [Google Scholar] [CrossRef]
- Sokol, N.S.; Ambros, V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005, 19, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int. Heart J. 2009, 50, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.B.; Huang, J.C.; Chen, Y.R.; Yao, J.N.; Cen, W.N.; Li, J.Y.; Jiang, Y.F.; Chen, G.; Li, S.H. Role of miR-1 expression in clear cell renal cell carcinoma (ccRCC): A bioinformatics study based on GEO, ArrayExpress microarrays and TCGA database. Pathol. Res. Pract. 2018, 214, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Fullaondo, A.; Lee, S.Y. Identification of putative miRNA involved in Drosophila melanogaster immune response. Dev. Comp. Immunol. 2012, 36, 267–273. [Google Scholar] [CrossRef]
- Shrinet, J.; Jain, S.; Jain, J.; Bhatnagar, R.K.; Sunil, S. Next generation sequencing reveals regulation of distinct Aedes microRNAs during chikungunya virus development. PLoS Negl. Trop. Dis. 2014, 8, e2616. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Zheng, P.M.; Tu, Z.J.; Chen, X.J. OL-032 identification of miRNAs in Aedes albopictus, and determination of their expression profiles during developmental stages and blood feeding using Solexa sequencing. Int. J. Infect. Dis. 2010, 14, S14–S15. [Google Scholar] [CrossRef]
- Yan, H.; Zhou, Y.; Liu, Y.; Deng, Y.; Chen, X. MiR-252 of the Asian tiger mosquito Aedes albopictus regulates dengue virus replication by suppressing the expression of the dengue virus envelope protein. J. Med. Virol. 2014, 86, 1428–1436. [Google Scholar] [CrossRef]
- Lim, D.H.; Lee, S.; Han, J.Y.; Choi, M.S.; Hong, J.S.; Seong, Y.; Kwon, Y.S.; Lee, Y.S. Ecdysone-responsive microRNA-252-5p controls the cell cycle by targeting Abi in Drosophila. FASEB J. 2018, 32, 4519–4533. [Google Scholar] [CrossRef]
- Liu, J.; Yao, W.; Yao, Y.; Du, X.; Zhou, J.; Ma, B.; Liu, H.; Li, Q.; Pan, Z. MiR-92a inhibits porcine ovarian granulosa cell apoptosis by targeting Smad7 gene. FEBS Lett. 2014, 588, 4497–4503. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, M.; Qin, G.; Weintraub, N.L.; Tang, Y. MiR-92a regulates viability and angiogenesis of endothelial cells under oxidative stress. Biochem. Biophys. Res. Commun. 2014, 446, 952–958. [Google Scholar] [CrossRef]
- Kurze, C.; Le, C.Y.; Dussaubat, C.; Erler, S.; Kryger, P.; Lewkowski, O.; Müller, T.; Widder, M.; Moritz, R.F. Nosema tolerant honeybees (Apis mellifera) escape parasitic manipulation of apoptosis. PLoS ONE 2015, 10, e0140174. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Wang, S.; Xue, R.; Cao, G.; Hu, X.; Huang, M.; Zhang, Y.; Lu, Y.; Zhu, L.; Chen, F.; et al. The gene expression profile of resistant and susceptible Bombyx mori strains reveals cypovirus-associated variations in host gene transcript levels. Appl. Microbiol. Biotechnol. 2015, 99, 5175–5187. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.E.; Clem, R.J. Lack of involvement of haemocytes in the establishment and spread of infection in Spodoptera frugiperda larvae infected with the baculovirus Autographa californica M nucleopolyhedrovirus by intrahaemocoelic injection. J. Gen. Virol. 2002, 83, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.P.; Wu, X.F.; Gokulamma, K. Antiviral activity in the mulberry silkworm, Bombyx mori L. J. Zhejiang Univ. SCIENCE A 2006, 7, 350–356. [Google Scholar] [CrossRef]
- Higes, M.; Juarranz, Á.; Dias-Almeida, J.; Lucena, S.; Botías, C.; Meana, A.; García-Palencia, P.; Martín-Hernández, R. Apoptosis in the pathogenesis of Nosema ceranae (Microsporidia: Nosematidae) in honey bees (Apis mellifera). Environ. Microbiol. Rep. 2013, 5, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lai, C.; Gu, H.; Zhao, L.; Xia, M.; Yang, P.; Wang, X. MiR-194 inhibits innate antiviral immunity by targeting FGF2 in influenza H1N1 virus infection. Front. Microbiol. 2017, 8, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Yang, L.; Han, L.L.; Yue, B. MicroRNA-194 regulates lipopolysaccharide-induced cell viability by inactivation of nuclear factor-κB pathway. Cell. Physiol. Biochem. 2017, 43, 2470–2478. [Google Scholar] [CrossRef]
- Xie, W.; Huang, A.; Li, H.; Feng, L.Z.; Zhang, F.P.; Guo, W.S. Identification and comparative analysis of microRNAs in Pinus massoniana, infected by Bursaphelenchus xylophilus. Plant Growth Regul. 2016, 1–10. [Google Scholar] [CrossRef]
- Saravanan, S.; Islam, V.I.; Thirugnanasambantham, K.; Sekar, D. In silico identification of human miR 3654 and its targets revealed its involvement in prostate cancer progression. Microrna 2016, 5, 1–6. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Tags |
|---|---|---|---|
| AcCK1-1 | 12,757,706 | 12,049,987 (94.45%) | 10,213,625 (84.76%) |
| AcCK1-2 | 12,794,543 | 12,122,624 (94.75%) | 10,275,851 (84.77%) |
| AcCK1-3 | 11,328,883 | 10,727,931 (94.70%) | 8,671,328 (80.83%) |
| AcCK2-1 | 17,161,911 | 16,292,122 (94.93%) | 14,528,074 (89.17%) |
| AcCK2-2 | 11,666,305 | 11,306,117 (96.91%) | 9,862,922 (87.24%) |
| AcCK2-3 | 11,757,223 | 11,328,016 (96.35%) | 9,310,382 (82.19%) |
| AcT1-1 | 11,213,906 | 10,923,950 (97.41%) | 9,356,024 (85.65%) |
| AcT1-2 | 14,549,245 | 13,778,004 (94.70%) | 11,661,491 (84.64%) |
| AcT1-3 | 11,029,767 | 10,800,311 (97.92%) | 8,914,299 (82.54%) |
| AcT2-1 | 13,263,930 | 12,775,381 (96.32%) | 10,974,972 (85.91%) |
| AcT2-2 | 13,688,082 | 13,150,156 (96.07%) | 10,977,135 (83.48%) |
| AcT2-3 | 12,936,357 | 12,467,737 (96.38%) | 10,859,224 (87.10%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Du, Y.; Chen, H.; Fan, Y.; Fan, X.; Zhu, Z.; Wang, J.; Xiong, C.; Zheng, Y.; Hou, C.; et al. Comparative Identification of MicroRNAs in Apis cerana cerana Workers’ Midguts in Response to Nosema ceranae Invasion. Insects 2019, 10, 258. https://doi.org/10.3390/insects10090258
Chen D, Du Y, Chen H, Fan Y, Fan X, Zhu Z, Wang J, Xiong C, Zheng Y, Hou C, et al. Comparative Identification of MicroRNAs in Apis cerana cerana Workers’ Midguts in Response to Nosema ceranae Invasion. Insects. 2019; 10(9):258. https://doi.org/10.3390/insects10090258
Chicago/Turabian StyleChen, Dafu, Yu Du, Huazhi Chen, Yuanchan Fan, Xiaoxue Fan, Zhiwei Zhu, Jie Wang, Cuiling Xiong, Yanzhen Zheng, Chunsheng Hou, and et al. 2019. "Comparative Identification of MicroRNAs in Apis cerana cerana Workers’ Midguts in Response to Nosema ceranae Invasion" Insects 10, no. 9: 258. https://doi.org/10.3390/insects10090258
APA StyleChen, D., Du, Y., Chen, H., Fan, Y., Fan, X., Zhu, Z., Wang, J., Xiong, C., Zheng, Y., Hou, C., Diao, Q., & Guo, R. (2019). Comparative Identification of MicroRNAs in Apis cerana cerana Workers’ Midguts in Response to Nosema ceranae Invasion. Insects, 10(9), 258. https://doi.org/10.3390/insects10090258