Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection


Abstract
1. Introduction
2. Materials and Methods
2.1. N. ceranae Spore Purification
2.2. Experimental Design and Sample Collection
2.3. Mortality Rate Analysis
2.4. RNA Extraction, Strand-Specific cDNA Library Construction and Deep Sequencing
2.5. Quality Control and Mapping of Reads
2.6. Transcripts Assembly
2.7. Bioinformatic Pipeline for Identification and Annotation of lncRNAs, and Quantification
2.8. DElncRNAs, Target Gene, and ceRNA Analyses
2.9. Real-time quantitative PCR (RT-qPCR) confirmation of DElncRNAs
2.10. Statistical Analysis
3. Results
3.1. The Effect of N. ceranae Inoculation Dose on the Mortality of A. m. ligustica Workers
3.2. Sequencing Results and Quality Control
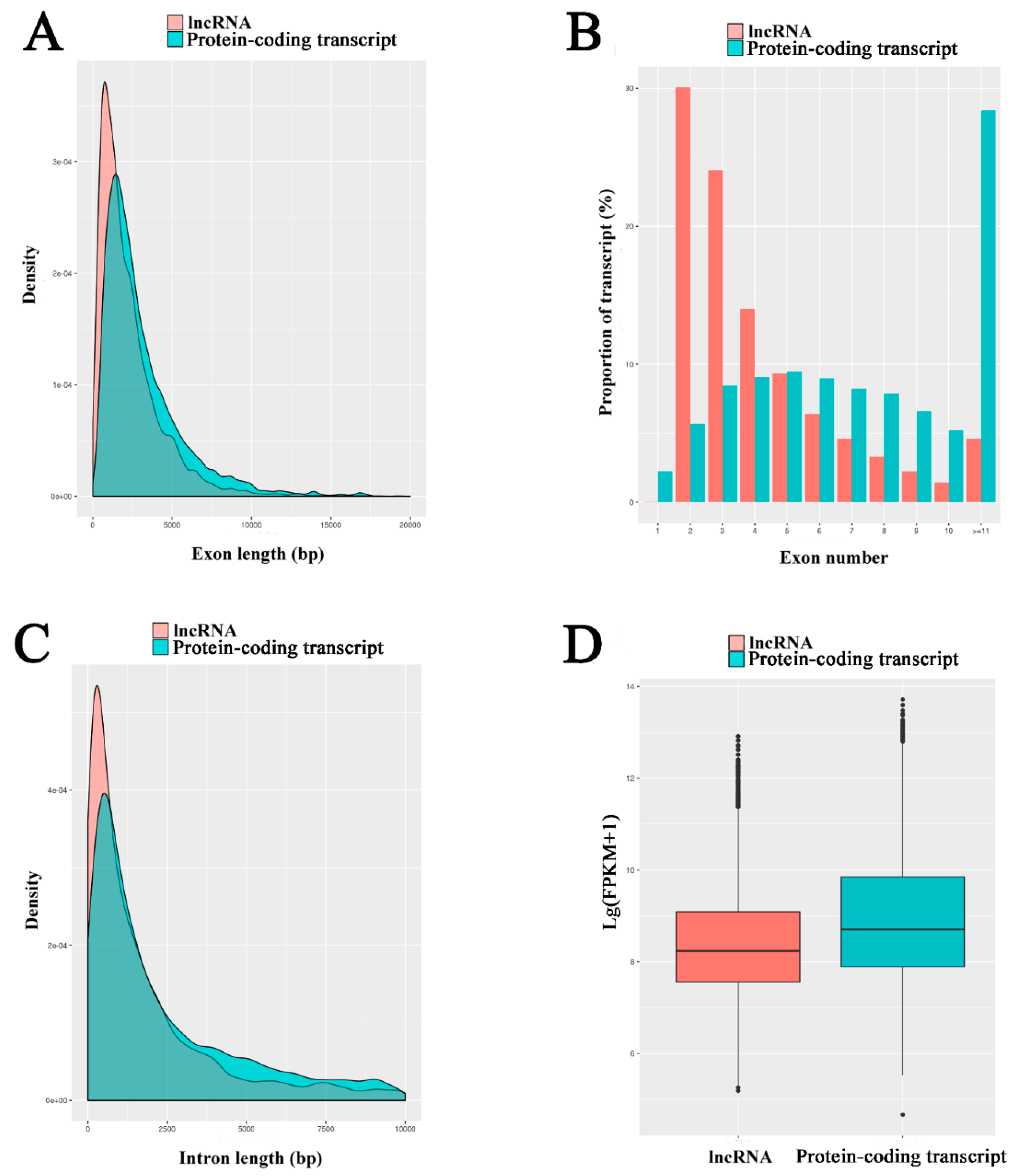
3.3. Characterization and Validation of A. m. ligustica lncRNAs
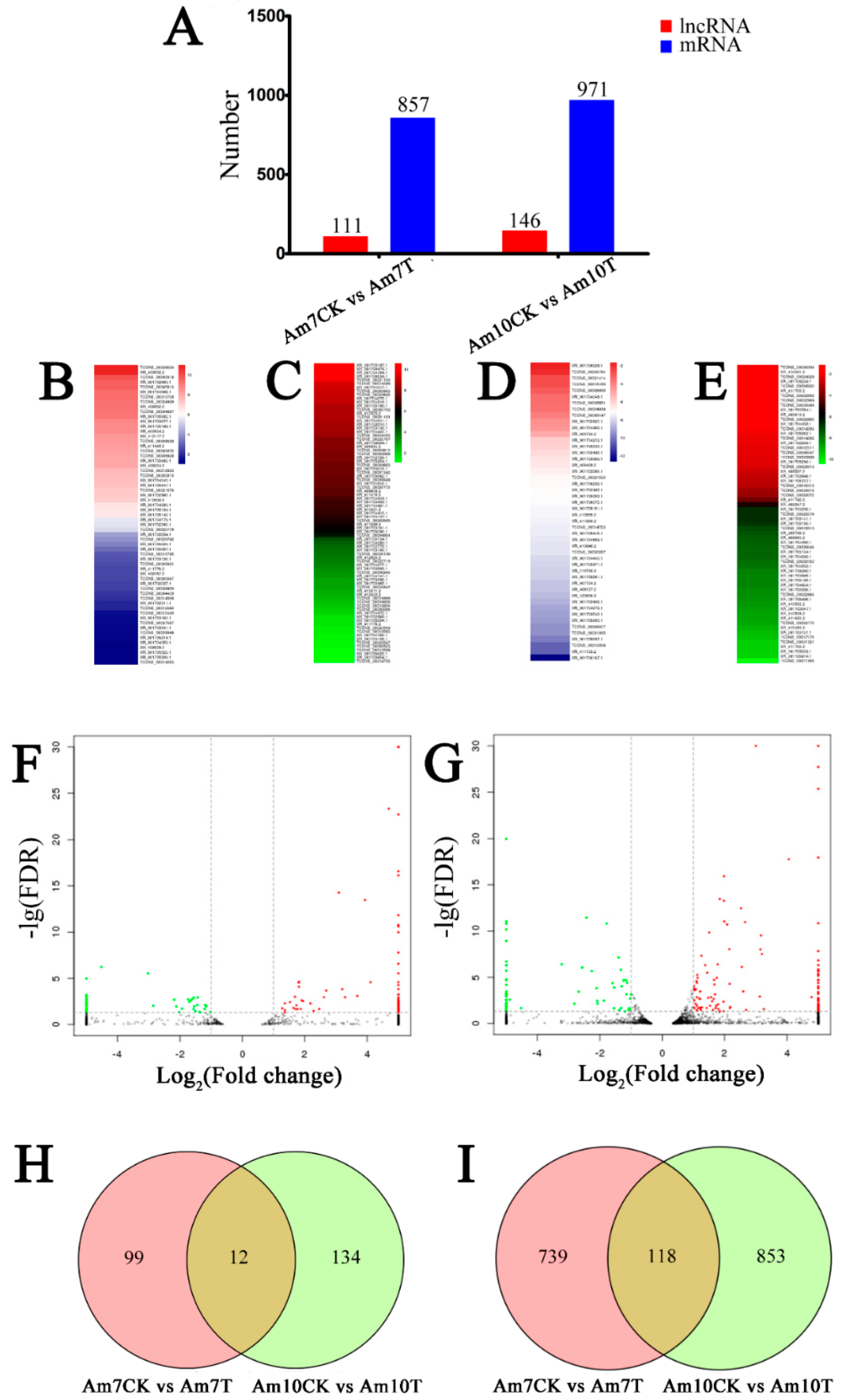
3.4. Identification of A. m. ligustica lncRNAs that Respond to N. ceranae Stress
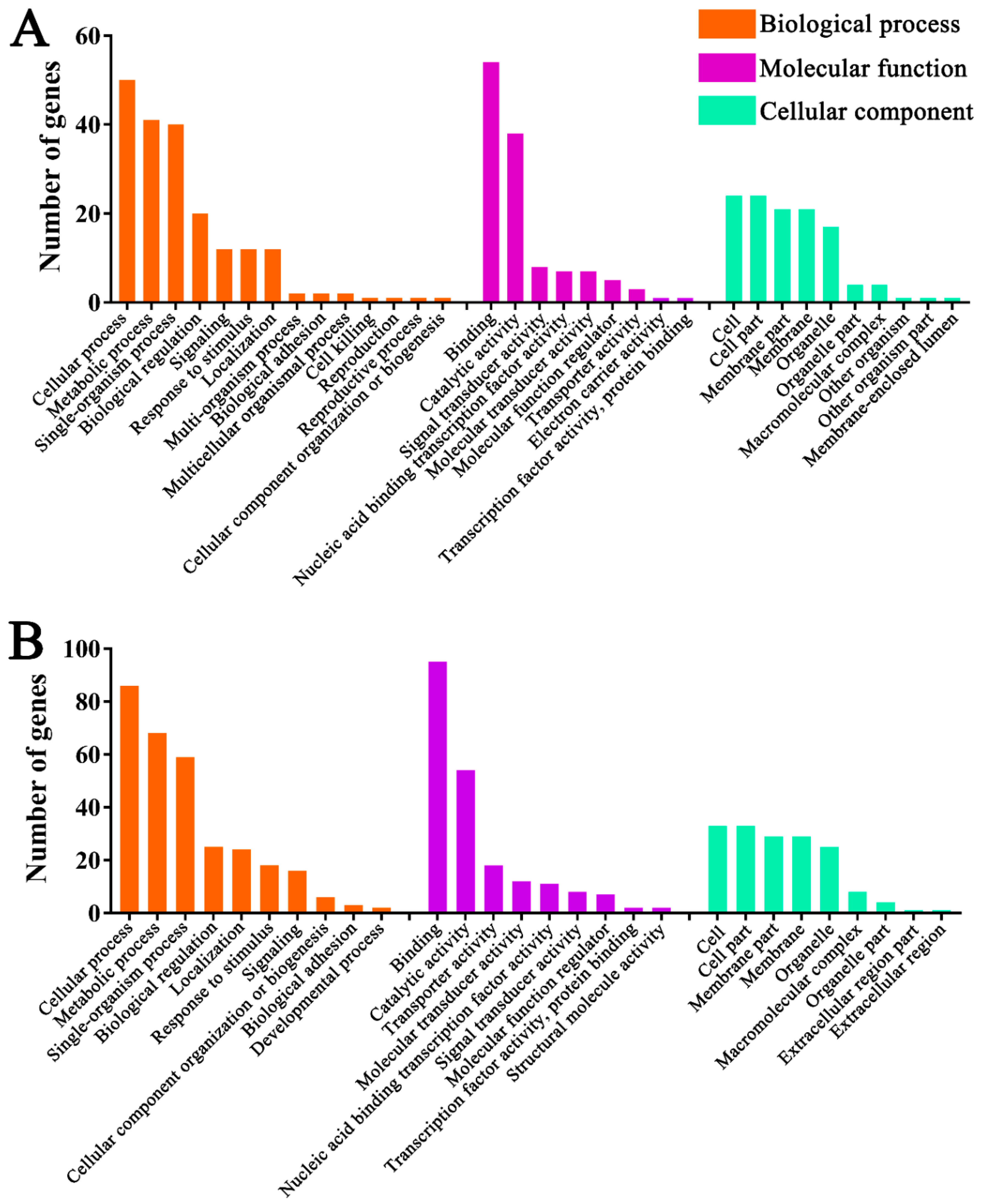
3.5. Functional Investigation of N. ceranae-Responsive lncRNAs in A. m. ligustica Workers’ Midguts
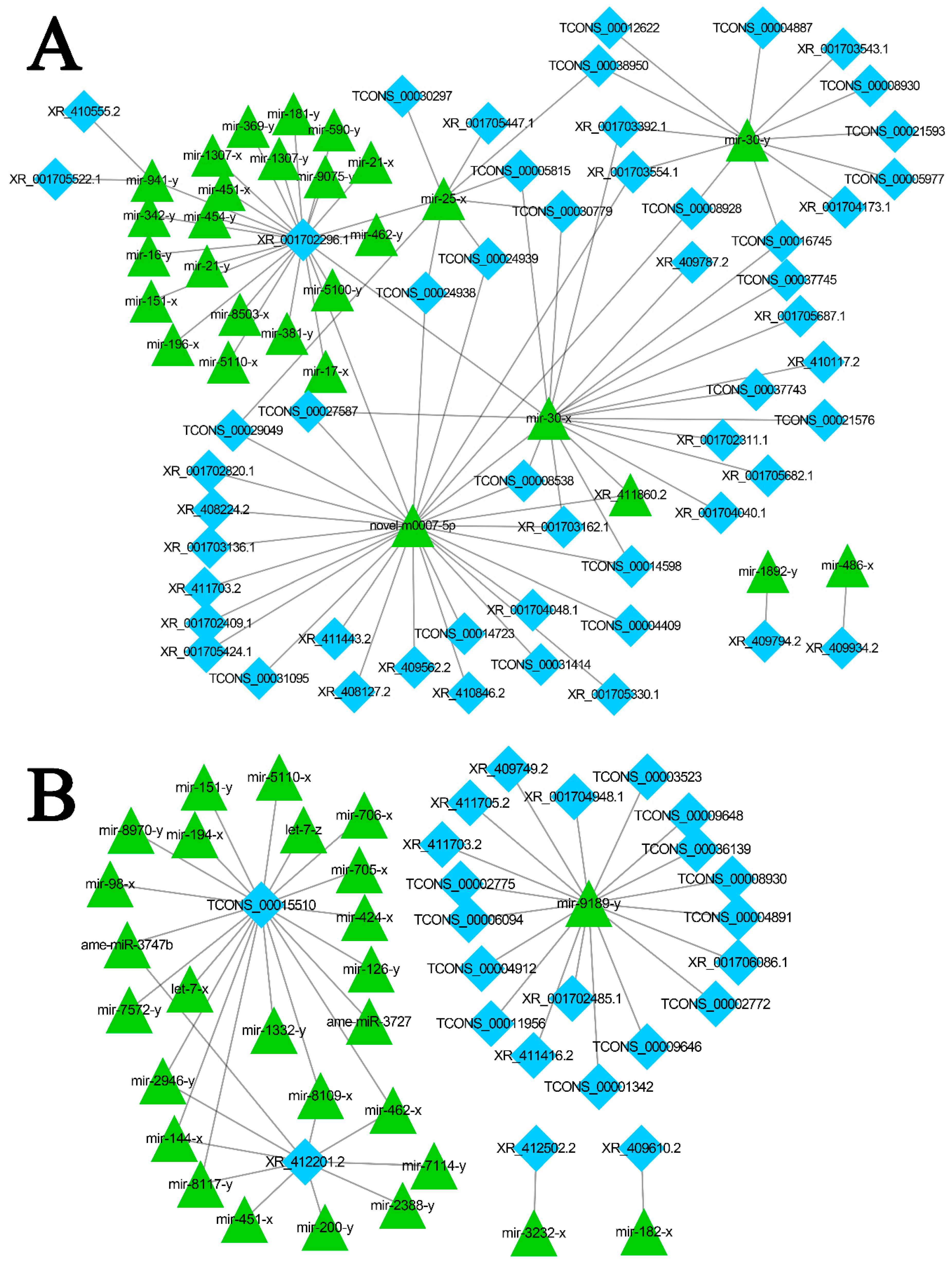
3.6. Discovery of A. m. ligustica lncRNAs as miRNA Precursors and ceRNAs
3.7. DElncRNA-miRNA-mRNA Regulatory Networks in A. m. ligustica Workers’ Midguts Invaded by N. ceranae
3.8. Validation of DElncRNAs byRT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bromenshenk, J.J.; Henderson, C.B.; Seccomb, R.A.; Welch, P.M.; Debnam, S.E.; Firth, D.R. Bees as biosensors: Chemosensory ability, honey bee monitoring systems, and emergent sensor technologies derived from the pollinator syndrome. Biosensors 2015, 5, 678–711. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis Mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Simpson, A.G.; Farmer, M.A.; Andersen, R.A.; Anderson, O.R.; Barta, J.R.; Bowser, S.S.; Brugerolle, G.; Fensome, R.A.; Fredericq, S.; et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot. Microbiol. 2005, 52, 399–451. [Google Scholar] [CrossRef] [PubMed]
- Szumowski, S.C.; Troemel, E.R. Microsporidia-host interactions. Curr. Opin. Microbiol. 2015, 26, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Visvesvara, G.S. In vitro cultivation of microsporidia of clinical importance. Clin. Microbiol. Rev. 2002, 15, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Evans, J.D.; Murphy, C.; Gutell, R.; Zuker, M.; Gundensen-Rindal, D.; Pettis, J.S. Morphological, molecular, and phylogenetic characterization of Nosema ceranae, a microsporidian parasite isolated from the European honey bee, Apis Mellifera. J. Eukaryot. Microbiol. 2009, 56, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Fries, I.; Feng, F.; da Silva, A.; Slemenda, S.B.; Pieniazek, N.J. Nosema ceranae n. sp. (Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (Hymenoptera, Apidae). Eur. J. Protistol. 1996, 32, 356–365. [Google Scholar] [CrossRef]
- Higes, M.; Martin, R.; Meana, A. Nosema ceranae, a new microsporidian parasite in honeybees in Europe. J. Invertebr. Pathol. 2006, 92, 93–95. [Google Scholar] [CrossRef]
- Huang, W.F.; Jiang, J.H.; Chen, Y.W.; Wang, C.H. A Nosema ceranae isolate from the honeybee Apis Mellifera. Apidologie 2007, 38, 30–37. [Google Scholar] [CrossRef]
- Klee, J.; Besana, A.M.; Genersch, E.; Gisder, S.; Nanetti, A.; Tam, D.Q.; Chinh, T.X.; Puerta, F.; Ruz, J.M.; Kryger, P.; et al. Widespread dispersal of the microsporidian Nosema ceranae, an emergent pathogen of the western honey bee, Apis mellifera. J. Invertebr. Pathol. 2007, 96, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Evans, J.D.; Smith, I.B.; Pettis, J.S. Nosema ceranae is a long-present and wide-spread microsporidian infection of the European honey bee (Apis mellifera) in the United States. J. Invertebr. Pathol. 2008, 97, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Botias, C.; Martin-Hernandez, R.; Dias, J.; Garcia-Palencia, P.; Matabuena, M.; Juarranz, A.; Barrios, L.; Meana, A.; Nanetti, A.; Higes, M. The effect of induced queen replacement on Nosema spp. infection in honey bee (Apis mellifera iberiensis) colonies. Environ. Microbiol. 2012, 14, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Pettis, J.S.; vanEngelsdorp, D.; Johnson, J.; Dively, G. Pesticide exposure in honey bees results in increased levels of the gut pathogen Nosema. Die Nat. 2012, 99, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Aufauvre, J.; Biron, D.G.; Vidau, C.; Fontbonne, R.; Roudel, M.; Diogon, M.; Vigues, B.; Belzunces, L.P.; Delbac, F.; Blot, N. Parasite-insecticide interactions: A case study of Nosema ceranae and fipronil synergy on honeybee. Sci. Rep. 2012, 2, 326. [Google Scholar] [CrossRef]
- Chen, G.; Qiu, C.; Zhang, Q.; Liu, B.; Cui, Q. Genome-wide analysis of human SNPs at long intergenic noncoding RNAs. Hum. Mutat. 2013, 34, 338–344. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes. Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Wang, M.B. Molecular functions of long non-coding RNAs in plants. Genes 2012, 3, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Yedavalli, V.R.; Niu, X.; Jerebtsova, M.; Van Eynde, A.; Beullens, M.; Bollen, M.; Jeang, K.T.; Nekhai, S. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J. Biol. Chem. 2011, 286, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Kasukawa, T.; Oyama, R.; Gough, J.; Frith, M.; Engstrom, P.G.; Lenhard, B.; Aturaliya, R.N.; Batalov, S.; Beisel, K.W.; et al. Transcript annotation in FANTOM3: Mouse gene catalog based on physical cDNAs. PLoS Genet. 2006, 2, e62. [Google Scholar] [CrossRef] [PubMed]
- Khachane, A.N.; Harrison, P.M. Mining mammalian transcript data for functional long non-coding RNAs. PLoS ONE 2010, 5, e10316. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X.; Stolc, V.; Li, X.; Zhang, D.; Su, N.; Tongprasit, W.; Li, S.; Cheng, Z.; Wang, J.; et al. Genome-wide transcription analyses in rice using tiling microarrays. Nat. Genet. 2006, 38, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef]
- Ben Amor, B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, D.F.; Xiong, C.L.; Hou, C.S.; Zheng, Y.Z.; Fu, Z.M.; Diao, Q.Y.; Zhang, L.; Wang, H.P.; Hou, Z.X.; et al. Identification of long non-coding RNAs in the chalkbrood disease pathogen Ascospheara Apis. J. Invertebr. Pathol. 2018, 156, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, D.F.; Xiong, C.L.; Hou, C.S.; Zheng, Y.Z.; Fu, Z.M.; Liang, Q.; Diao, Q.Y.; Zhang, L.; Wang, H.P.; et al. First identification of long non-coding RNAs in fungal parasite Nosema ceranae. Apidologie 2018, 49, 660–670. [Google Scholar] [CrossRef]
- Etebari, K.; Furlong, M.J.; Asgari, S. Genome wide discovery of long intergenic non-coding RNAs in Diamondback moth (Plutella xylostella) and their expression in insecticide resistant strains. Sci. Rep. 2015, 5, 14642. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.M.; Waterhouse, R.M.; Muskavitch, M.A. Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genom. 2015, 16, 337. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [PubMed]
- Humann, F.C.; Tiberio, G.J.; Hartfelder, K. Sequence and expression characteristics of long noncoding RNAs in honey bee caste development–potential novel regulators for transgressive ovary size. PLoS ONE 2013, 8, e78915. [Google Scholar] [CrossRef]
- Sawata, M.; Yoshino, D.; Takeuchi, H.; Kamikouchi, A.; Ohashi, K.; Kubo, T. Identification and punctate nuclear localization of a novel noncoding RNA, Ks-1, from the honeybee brain. RNA 2002, 8, 772–785. [Google Scholar] [CrossRef]
- Jayakodi, M.; Jung, J.W.; Park, D.; Ahn, Y.J.; Lee, S.C.; Shin, S.Y.; Shin, C.; Yang, T.J.; Kwon, H.W. Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey bees Apis cerana and Apis mellifera. BMC Genom. 2015, 16, 680. [Google Scholar] [CrossRef]
- Chen, X.; Ma, C.; Chen, C.; Lu, Q.; Shi, W.; Liu, Z.; Wang, H.; Guo, H. Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees. Peer J. 2017, 5, e3881. [Google Scholar] [CrossRef]
- Cornman, R.S.; Chen, Y.P.; Schatz, M.C.; Street, C.; Zhao, Y.; Desany, B.; Egholm, M.; Hutchison, S.; Pettis, J.S.; Lipkin, W.I.; et al. Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog. 2009, 5, e1000466. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E. Development of a rapid and sensitive RT-PCR method for the detection of deformed wing virus, a pathogen of the honeybee (Apis mellifera). Vet. J. 2005, 169, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.; Shen, X.R.; Boggis, C.; Sisson, G. Molecular diagnosis of Kashmir bee virus infection. J. Apic. Res. 1995, 34, 153–160. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA viruses in hymenopteran pollinators: Evidence of inter-Taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef] [PubMed]
- Ribiere, M.; Triboulot, C.; Mathieu, L.; Aurieres, C.; Faucon, J.P.; Pepin, M. Molecular diagnosis of chronic bee paralysis virus infection. Apidologie 2002, 33, 339–351. [Google Scholar] [CrossRef]
- Benjeddou, M.; Leat, N.; Allsopp, M.; Davison, S. Detection of acute bee paralysis virus and black queen cell virus from honeybees by reverse transcriptase pcr. Appl. Env. Microbiol. 2001, 67, 2384–2387. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, C.; Evans, J.D.; Li, W.; Branchiccela, B.; Li, J.H.; Heerman, M.C.; Banmeke, O.; Zhao, Y.; Hamilton, M.; Higes, M.; et al. Nosemosis control in European honey bees, Apis mellifera, by silencing the gene encoding Nosema ceranae polar tube protein 3. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef]
- Meana, A.; Martín-Hernández, R.; Higes, M. The reliability of spore counts to diagnose Nosema ceranae infections in honey bees. J. Apic. Res. 2010, 49, 212–214. [Google Scholar] [CrossRef]
- Forsgren, E.; Fries, I. Comparative virulence of Nosema ceranae and Nosema apis in individual European honey bees. Vet. Parasitol. 2010, 170, 212–217. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, Y.P.; Wang, R.W.; Cheng, S.; Evans, J.D. Host-parasite interactions and purifying selection in a microsporidian parasite of honey bees. PLoS ONE 2016, 11, e0147549. [Google Scholar] [CrossRef]
- Huang, W.F.; Solter, L.F. Comparative development and tissue tropism of Nosema apis and Nosema ceranae. J. Invertebr. Pathol. 2013, 113, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Antonio Gomez, J.L.; Wapinski, O.W.; Yang, Y.; Bureau, J.F.; Gopinath, S.; Monack, D.Y.; Chang, H.; Brahic, M.; Kirkegaard, K. The nest long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, W. microRNA Target Prediction. Methods Mol. Biol. 2017, 1513, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dussaubat, C.; Brunet, J.L.; Higes, M.; Colbourne, J.K.; Lopez, J.; Choi, J.H.; Martin-Hernandez, R.; Botias, C.; Cousin, M.; McDonnell, C.; et al. Gut pathology and responses to the microsporidium Nosema ceranae in the honey bee Apis mellifera. PLoS ONE 2012, 7, e37017. [Google Scholar] [CrossRef] [PubMed]
- Higes, M.; Garcia-Palencia, P.; Martin-Hernandez, R.; Meana, A. Experimental infection of Apis mellifera honeybees with Nosema ceranae (Microsporidia). J. Invertebr. Pathol. 2007, 94, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. 2016, 6, 20238. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Han, K.; Zhang, G.; Wang, J.; Xie, K.; Xue, Q. Genome-wide analysis of lncRNA and mRNA expression during differentiation of abdominal preadipocytes in the Chicken. Genes Genomes Genet. 2017, 7, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Z.; Zhang, B.; Yu, Q.Y.; Zhang, Z. BmncRNAdb: A comprehensive database of non-coding RNAs in the silkworm, Bombyx mori. BMC Bioinform. 2016, 17, 370. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, Z.; Bailey, T.L.; Perkins, A.C.; Tallack, M.R.; Xu, Z.; Liu, H. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC Bioinform. 2012, 13, 331. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; Yamazaki, T.; Hirose, T. Architectural RNAs (arcRNAs): A class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim. Biophys. Acta 2016, 1859, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Shu, S.; Cheng, H. A novel lncRNA-mediated trans-regulatory mechanism in the development of cleft palate in mouse. Mol. Genet. Genom. Med. 2019, 7, e00522. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Gong, J.; Liu, W.; Zhang, J.; Miao, X.; Guo, A.Y. lncRNASNP: A database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. 2015, 43, D181–D186. [Google Scholar] [CrossRef]
- Liu, K.; Yan, Z.; Li, Y.; Sun, Z. Linc2GO: A human LincRNA function annotation resource based on ceRNA hypothesis. Bioinformatics 2013, 29, 2221–2222. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Boley, N.; Eisman, R.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the Drosophila transcriptome. Nature 2014, 512, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Yuan, Z.; Guo, D.; Hou, B.; Yin, C.; Zhang, W.; Li, F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genom. 2015, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- Holt, H.L.; Aronstein, K.A.; Grozinger, C.M. Chronic parasitization by Nosema microsporidia causes global expression changes in core nutritional, metabolic and behavioral pathways in honey bee workers (Apis mellifera). BMC Genom. 2013, 14, 799. [Google Scholar] [CrossRef] [PubMed]
- Badaoui, B.; Fougeroux, A.; Petit, F.; Anselmo, A.; Gorni, C.; Cucurachi, M.; Cersini, A.; Granato, A.; Cardeti, G.; Formato, G.; et al. RNA-sequence analysis of gene expression from honeybees (Apis mellifera) infected with Nosema ceranae. PLoS ONE 2017, 12, e0173438. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, Y.; Wang, R.W.; Schwarz, R.S.; Evans, J.D. Honey bee microRNAs respond to infection by the microsporidian parasite Nosema ceranae. Sci. Rep. 2015, 5, 17494. [Google Scholar] [CrossRef]
- Huang, Q.; Evans, J.D. Identification of microRNA-like small RNAs from fungal parasite Nosema ceranae. J. Invertebr. Pathol. 2016, 133, 107–109. [Google Scholar] [CrossRef]
- Evans, J.D.; Huang, Q. Interactions among host-parasite microRNAs during Nosema ceranae proliferation in Apis mellifera. Front. Microbiol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.; Chen, H.; Xiong, C.; Zheng, Y.; Hou, C.; Du, Y.; Geng, S.; Wang, H.; Dingding, Z.; et al. Genome-wide identification of circular rnas in fungal parasite Nosema ceranae. Curr. Microbiol. 2018, 75, 1655–1660. [Google Scholar] [CrossRef]
- Broadbent, K.M.; Broadbent, J.C.; Ribacke, U.; Wirth, D.; Rinn, J.; Sabeti, P. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genom. 2015, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Briskine, R.; Hirsch, C.N.; Myers, C.L.; Springer, N.M.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS ONE 2013, 8, e61005. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Bruning, J.C. Regulation of metabolism by long, non-coding RNAs. Front. Genet. 2014, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, S.; Wu, R.; Zhou, X.; Zhu, D.; Zhang, Y. Identification of long non-protein coding RNAs in chicken skeletal muscle using next generation sequencing. Genomics 2012, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, G.; Chen, L.; Jiang, J.; Liu, L.; Li, N.; Zhao, J.; Sun, X.; Zhou, P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genom. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Weikard, R.; Hadlich, F.; Kuehn, C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genom. 2013, 14, 789. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Young, R.S.; Marques, A.C.; Tibbit, C.; Haerty, W.; Bassett, A.R.; Liu, J.L.; Ponting, C.P. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol. Evol. 2012, 4, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ding, Y.; Zhan, F.; Zhang, H.; Han, B.; Hu, G.; Zhao, K.; Yang, N.; Yu, Y.; Mao, L.; et al. The conservation and signatures of lincRNAs in Marek’s disease of chicken. Sci. Rep. 2015, 5, 15184. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-kappaB activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, A.; Uhl, M.; Zhang, Y.; Lin, Y.Y.; Niknafs, Y.R.; Ramnarine, V.; Malik, R.; Feng, F.M.; Chinnaiyan, A.; Collins, C.; et al. MechRNA: Prediction of lncRNA mechanisms from RNA-RNA and RNA-protein interactions. Bioinformatics 2018, 34. [Google Scholar] [CrossRef] [PubMed]
- Mayack, C.; Naug, D. Energetic stress in the honeybee Apis mellifera from Nosema ceranae infection. J. Invertebr. Pathol. 2009, 100, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Kurze, C.; Le Conte, Y.; Dussaubat, C.; Erler, S.; Kryger, P.; Lewkowski, O.; Muller, T.; Widder, M.; Moritz, R.F. Nosema tolerant honeybees (Apis mellifera) escape parasitic manipulation of apoptosis. PLoS ONE 2015, 10, e0140174. [Google Scholar] [CrossRef] [PubMed]
- Kurze, C.; Le Conte, Y.; Kryger, P.; Lewkowski, O.; Müller, T.; Moritz, R.F.A. Infection dynamics of Nosema ceranae in honey bee midgut and host cell apoptosis. J. Invertebr. Pathol. 2018, 154, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, R.; Higes, M.; Sagastume, S.; Juarranz, Á.; Dias-Almeida, J.; Budge, G.E.; Meana, A.; Boonham, N. Microsporidia infection impacts the host cell’s cycle and reduces host cell apoptosis. PLoS ONE 2017, 12, e0170183. [Google Scholar] [CrossRef] [PubMed]
- Doublet, V.; Poeschl, Y.; Gogol-Doring, A.; Alaux, C.; Annoscia, D.; Aurori, C.; Barribeau, S.M.; Bedoya-Reina, O.C.; Brown, M.J.; Bull, J.C.; et al. Unity in defence: Honeybee workers exhibit conserved molecular responses to diverse pathogens. BMC Genom. 2017, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Gliński, Z.; Jarosz, J. Infection and immunity in the honey bee Apis mellifera. Apiacta 2001, 36, 12–24. [Google Scholar] [CrossRef]
- Stroschein-Stevenson, S.L.; Foley, E.; O’Farrell, P.H.; Johnson, A.D. Phagocytosis of Candida albicans by RNAi-treated Drosophila S2 cells. Methods Mol. Biol. 2009, 470, 347–358. [Google Scholar] [CrossRef]
- Bonasio, R.; Shiekhattar, R. Regulation of transcription by long noncoding RNAs. Annual. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Moltke, I.; Roth, A.; Washietl, S.; Wen, J.; Kellis, M.; Breaker, R.; Pedersen, J.S. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 2011, 21, 1929–1943. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Somoza, I.; Weigel, D.; Franco-Zorilla, J.M.; Garcia, J.A.; Paz-Ares, J. ceRNAs: miRNA target mimic mimics. Cell 2011, 147, 1431–1432. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.F.; Zhong, Q.; Xia, T.L.; Chen, Q.; Zhang, M.Y.; Zhou, A.J.; Tu, Z.W.; Qu, C.; Li, M.Z.; Xia, Y.F.; et al. RBM24 suppresses cancer progression by upregulating miR-25 to target MALAT1 in nasopharyngeal carcinoma. Cell Death Dis. 2016, 7, e2352. [Google Scholar] [CrossRef]
- Xie, M.; Qin, H.; Luo, Q.; Huang, Q.; He, X.; Yang, Z.; Lan, P.; Lian, L. MicroRNA-30a regulates cell proliferation and tumor growth of colorectal cancer by targeting CD73. BMC Cancer 2017, 17, 305. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
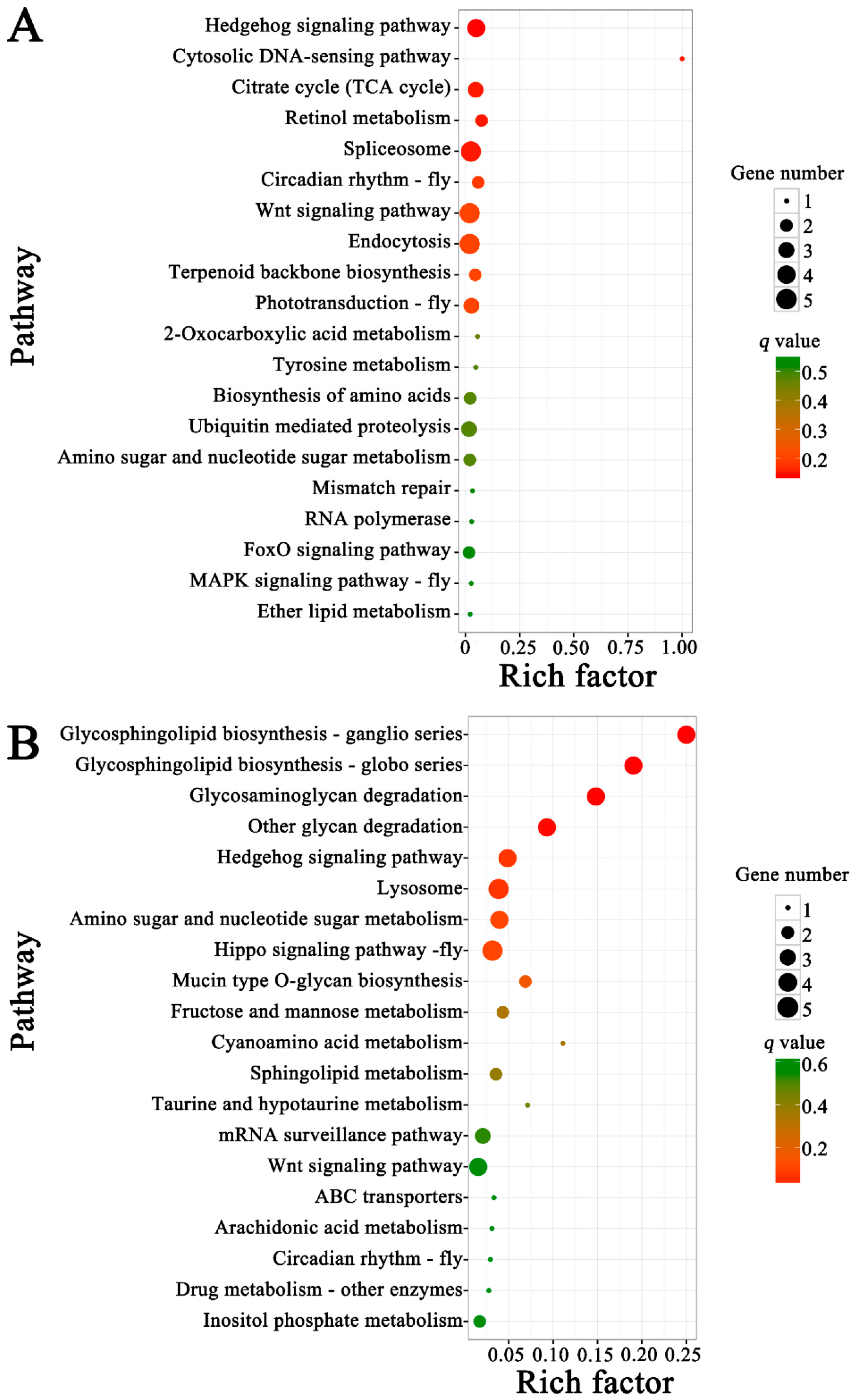{kind=link}
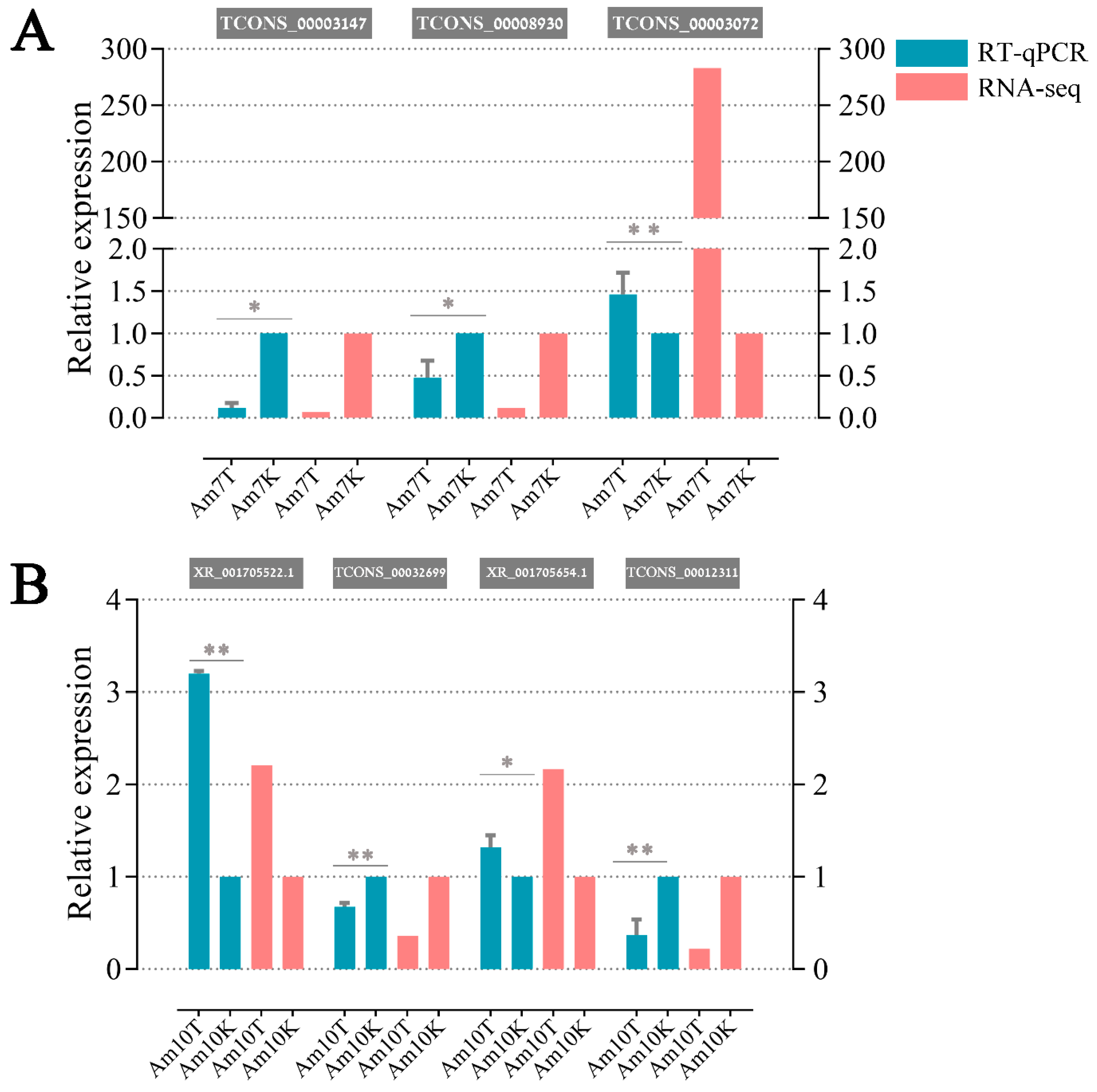{kind=link}
| Sample | Raw Reads | Clean Reads (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| Am7CK1 | 160,844,082 | 160,049,106 (99.51) | 23,340,144,349 (97.41) | 22,521,956,996 (94.00) |
| Am7CK2 | 129,878,194 | 129,283,918 (99.54) | 18,891,245,674 (97.56) | 18,239,412,915 (94.19) |
| Am7CK3 | 113,683,898 | 113,165,446 (99.54) | 16,535,666,991 (97.52) | 15,943,589,998 (94.03) |
| Am7T1 | 152,323,278 | 151,668,484 (99.57) | 22,161,043,664 (97.55) | 21,387,125,499 (94.15) |
| Am7T2 | 200,417,896 | 199,313,090 (99.45) | 28,948,504,448 (97.11) | 27,829,913,730 (93.35) |
| Am7T3 | 126,667,596 | 126,053,962 (99.52) | 18,386,919,122 (97.38) | 17,719,616,862 (93.85) |
| Am10CK1 | 160,537,248 | 159,765,346 (99.52) | 23,262,715,888 (97.27) | 22,443,038,732 (93.84) |
| Am10CK2 | 149,230,808 | 148,494,716 (99.51) | 21,633,348,548 (97.28) | 20,852,891,752 (93.77) |
| Am10CK3 | 131,386,354 | 130,619,802 (99.42) | 18,959,297,638 (96.98) | 18,248,516,385 (93.34) |
| Am10T1 | 249,473,666 | 248,333,982 (99.54) | 36,162,922,479 (97.32) | 34,857,597,435 (93.81) |
| Am10T2 | 208,589,832 | 207,574,770 (99.51) | 30,251,988,213 (97.34) | 29,139,831,253 (93.77) |
| Am10T3 | 173,097,006 | 172,166,682 (99.46) | 25,113,348,781 (97.38) | 24,175,449,594 (93.74) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Chen, H.; Du, Y.; Zhou, D.; Geng, S.; Wang, H.; Wan, J.; Xiong, C.; Zheng, Y.; Guo, R. Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection. Insects 2019, 10, 245. https://doi.org/10.3390/insects10080245
Chen D, Chen H, Du Y, Zhou D, Geng S, Wang H, Wan J, Xiong C, Zheng Y, Guo R. Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection. Insects. 2019; 10(8):245. https://doi.org/10.3390/insects10080245
Chicago/Turabian StyleChen, Dafu, Huazhi Chen, Yu Du, Dingding Zhou, Sihai Geng, Haipeng Wang, Jieqi Wan, Cuiling Xiong, Yanzhen Zheng, and Rui Guo. 2019. "Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection" Insects 10, no. 8: 245. https://doi.org/10.3390/insects10080245
APA StyleChen, D., Chen, H., Du, Y., Zhou, D., Geng, S., Wang, H., Wan, J., Xiong, C., Zheng, Y., & Guo, R. (2019). Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection. Insects, 10(8), 245. https://doi.org/10.3390/insects10080245