Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy

Abstract
1. Introduction
2. The DMD Gene and its Mutations in Dystrophinopathy
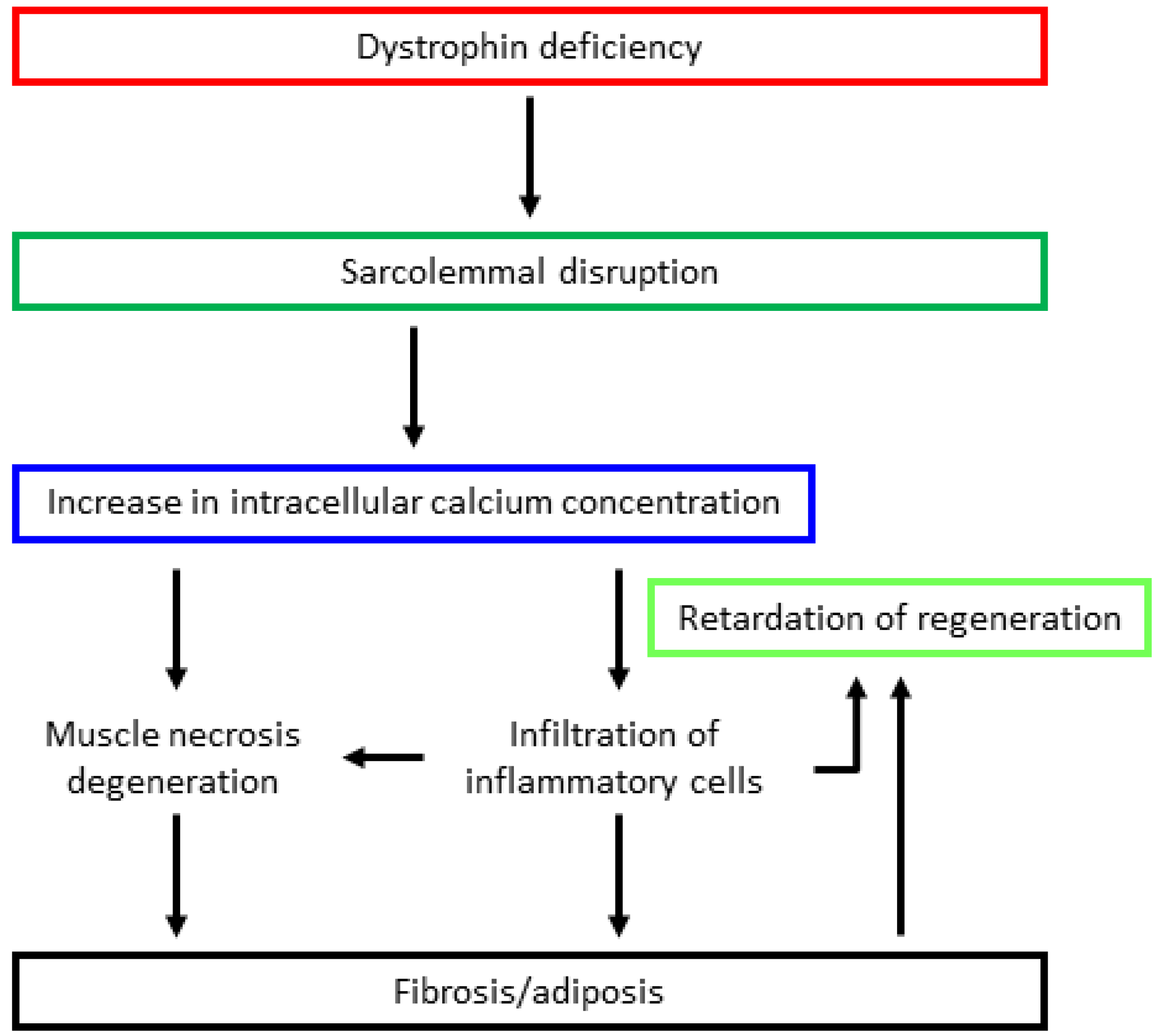
3. Pathomechanism of Dystrophinopathy
4. DMD Animal Models for the Development of Mutation-Specific Therapeutic Strategies
4.1. Mdx Mice
4.2. Mdx52 Mice
4.3. Dystrophic Dogs
5. Therapeutic Strategies Based on Mutations of DMD
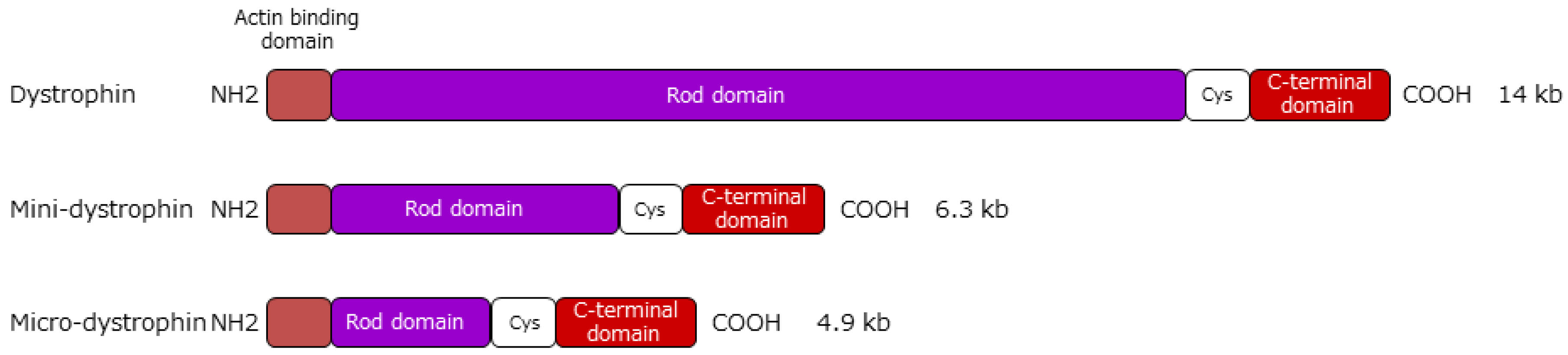
5.1. Gene Therapy Using Vectors
5.1.1. Lentiviral Vector
5.1.2. Adenoviral Vector
5.1.3. AAV Vector
5.1.4. HAC Vector
5.2. Readthrough Therapy
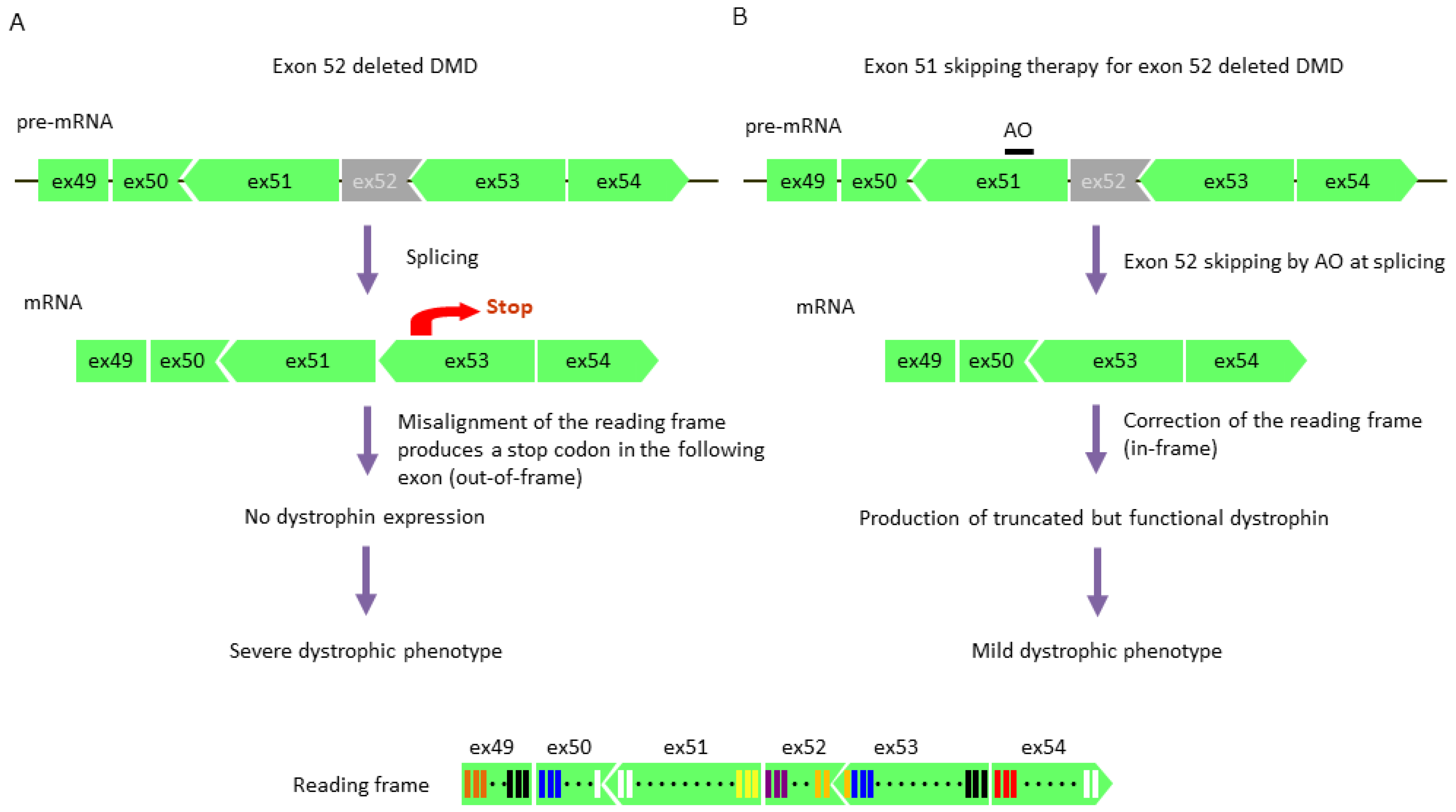
5.3. Exon Skipping Therapy
5.3.1. Exon Skipping Using Antisense Oligonucleotides
5.3.2. Preclinical Trials of Exon skipping Therapy using Antisense Oligonucleotides
5.3.3. Clinical Trials of Exon Skipping Therapy using Antisense Oligonucleotides
5.3.4. Issues and Prospects of Exon Skipping Therapy using AOs
5.3.5. Exon Skipping via Genome Editing
6. Future Aspects of Therapeutic Strategies
Funding
Conflicts of Interest
References
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker Muscular Dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Kesari, A.; Gordish-Dressman, H.; Cnaan, A.; Morgenroth, L.P.; Punetha, J.; Duong, T.; Henricson, E.K.; Pegoraro, E.; McDonald, C.M.; et al. Cooperative International Neuromuscular Research Group Investigators. Genetic modifiers of ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann. Neurol. 2015, 77, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Yoshida, M.; Nakamura, A.; Koyama, J.; Nanba, T.; Ohori, N.; Ikeda, S. Clinical characteristics of aged Becker muscular dystrophy patients with onset after 30 years. Eur. Neurol. 1999, 42, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, V.; Giliberto, F.; Muniz, G.M.; Francipane, L.; Marzese, D.M.; Mampel, A.; Roque, M.; Frechtel, G.D.; Szijan, I. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle Nerve 2009, 39, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Ikeda, S.; Nakamura, A.; Kagoshima, M.; Takeda, S.; Shoji, S.; Yanagisawa, N. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve 1993, 16, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Deepha, S.; Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Nalini, A.; Gayathri, N.; Purushottam, M. MLPA identification of dystrophin mutations and in silico evaluation of the predicted protein in dystrophinopathy cases from India. BMC Med. Genet. 2017, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Feng, J.; Buzin, C.H.; Scaringe, W.; Liu, Q.; Mendell, J.R.; den Dunnen, J.; Sommer, S.S. Three-tiered noninvasive diagnosis in 96% of patients with Duchenne muscular dystrophy (DMD). Hum. Mutat. 2004, 23, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Dent, K.M.; Dunn, D.M.; von Niederhausern, A.C.; Aoyagi, A.T.; Kerr, L.; Bromberg, M.B.; Hart, K.J.; Tuohy, T.; White, S.; den Dunnen, J.T.; et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am. J. Med. Genet. A 2005, 134, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Bridgeman, S.J. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J. Mol. Diagn. 2005, 7, 317–326. [Google Scholar]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- White, S.; Kalf, M.; Liu, Q.; Villerius, M.; Engelsma, D.; Kriek, M.; Vollebregt, E.; Bakker, B.; van Ommen, G.J.; Breuning, M.H.; et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 2002, 71, 365–374. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Aartsma-Rus, A.; Flanigan, K.M.; Weiss, R.B.; Kneppers, A.L.; Lalic, T.; Janson, A.A.; Ginjaar, H.B.; Breuning, M.H.; den Dunnen, J.T. Duplications in the DMD gene. Hum. Mutat. 2006, 7, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- van den Bergen, J.C.; Wokke, B.H.; Janson, A.A.; van Duinen, S.G.; Hulsker, M.A.; Ginjaar, H.B.; van Deutekom, J.C.; Aartsma-Rus, A.; Kan, H.E.; Verschuuren, J.J. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J. Neurol. Neurosurg. Psychiatry 2013, 85, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Raguenes-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Cheron, A.; Vie, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2014, 24, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotype differences between patients bearing partial deletions of the DMD locus. Genomics 1998, 2, 90–95. [Google Scholar] [CrossRef]
- Tuffery-Giraud, S.; Beroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Moizard, M.P.; Bernard, R.; Cossee, M.; Boisseau, P.; et al. Genotype-phenotype analysis in 2405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.R.; den Dunnen, J.; O’Brien, K.F.; Darras, B.T.; Kunkel, L.M. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet. 2001, 2, 17. [Google Scholar] [CrossRef]
- Mendell, J.R.; Buzin, C.H.; Feng, J.; Yan, J.; Serrano, C.; Sangani, D.S.; Wall, C.; Prior, T.W.; Sommer, S.S. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001, 57, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, L.C.; de Moura-Neto, R.S.; Falcao-Conceicao, D.N. DGGE analysis as a tool to identify point mutations, de novo mutations and carriers of the dystrophin gene. Neuromuscul. Disord. 2002, 12, 845–848. [Google Scholar] [CrossRef]
- Flanigan, K.M.; von Niederhausern, A.; Dunn, D.M.; Alder, J.; Mendell, J.R.; Weiss, R.B. Rapid direct sequence analysis of the dystrophin gene. Am. J. Hum. Genet. 2003, 72, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, R.M.; Mulder, I.M.; Vossen, R.; de Koning-Gansm, P.A.; Kraak, M.; Ginjaar, I.B.; van der Hout, A.H.; Bakker, E.; Buys, C.H.; van Ommen, G.J.; et al. DGGE-based whole-gene mutation scanning of the dystrophin gene in Duchenne and Becker muscular dystrophy patients. Hum. Mutat. 2004, 23, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loike, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L.; et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Urion, D.K.; Ghosh, P.S. Dystrophinopathies. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2018. [Google Scholar]
- Wong, L.J.; Dimmock, D.; Geraghty, M.T.; Quan, R.; Lichter-Konecki, U.; Wang, J.; Brundage, E.K.; Scaglia, F.; Chinault, A.C. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin. Chem. 2008, 54, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Takeda, S.; Lu, Q.L.; Partridge, T.A.; Nakamura, A.; Hoffman, E.P. A renaissance for antisense oligonucleotide drugs in neurology: Exon skipping breaks new ground. Arch. Neurol. 2009, 66, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Takeda, S. Current challenges and future directions in recombinant AAV-mediated gene therapy of Duchenne muscular dystrophy. Pharmaceuticals 2013, 6, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, M.; Fukuda, S.; Uezumi, A.; Masuda, S.; Miyoshi, H.; Yamamoto, H.; Wada, M.R.; Masubuchi, N.; Miyagoe-Suzuki, Y.; Takeda, S. Autologous transplantation of SM/C-2.6(+) satellite cells transduced with micro-dystrophin CS1 cDNA by lentiviral vector into mdx mice. Mol. Ther. 2007, 15, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Sunada, Y.; Campbell, K.P. Dystrophin-glycoprotein complex: Molecular organization and critical roles in skeletal muscle. Curr. Opin. Neurol. 1995, 8, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Worton, R. Muscular dystrophies: Diseases of the dystrophin-glycoprotein complex. Science 1995, 270, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Hutter, O.F.; Burton, F.L.; Bovell, D.L. Mechanical properties of normal and mdx mouse sarcolemma: Bearing on function of dystrophin. J. Muscle. Res. Cell Motil. 1991, 12, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+] i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappa B pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Sato, K.; Yokota, T.; Ichioka, S.; Shibata, M.; Takeda, S. Vasodilation of intramuscular arterioles under shear stress in dystrophin-deficient skeletal muscle is impaired through decreased nNOS expression. Acta Myol. 2008, 27, 30–36. [Google Scholar] [PubMed]
- Wakayama, Y.; Jimi, T.; Inoue, M.; Kojima, H.; Murahashi, M.; Kumagai, T.; Yamashita, S.; Hara, H.; Shibuya, S. Reduced aquaporin 4 expression in the muscle plasma membrane of patients with Duchenne muscular dystrophy. Arch. Neurol. 2002, 59, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Frigeri, A.; Nicchia, G.P.; Balena, R.; Nico, B.; Svelto, M. Aquaporins in skeletal muscle: Reassessment of the functional role of aquaporin-4. FASEB J. 2004, 18, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Hirn, C.; Shapovalov, G.; Petermann, O.; Roulet, E.; Ruegg, U.T. Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 2008, 132, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, O.; von Wegner, F.; Chamberlain, J.S.; Fink, R.H.; Rohrbach, P. L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle. PLoS ONE 2008, 3, e1762. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Komamura, K.; Miyatake, K.; Shigekawa, M. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J. Cell Biol. 2003, 161, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, C.Y.; Taniguti, A.P.; Pertille, A.; Santo Neto, H.; Marques, M.J. Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am. J. Physiol. Cell Physiol. 2011, 301, C1344–C1350. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.A.; Zheng, R.; Gonzalez, J.P.; Xie, L.H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric oxide signaling pathway in Duchenne muscular dystrophy mice: Up-regulation of L-arginine transporters. Biochem. J. 2013, 449, 13342. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Clementi, E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol. Neurobiol. 2012, 46, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Ye, J.; De Nardi, C.; Monopoli, A.; Ongini, E.; Victor, R.G. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS ONE 2012, 7, e49350. [Google Scholar] [CrossRef] [PubMed]
- Mizunoya, W.; Upadhaya, R.; Burczynski, F.J.; Wang, G.; Anderson, J.E. Nitric oxide donors improve prednisone effects on muscular dystrophy in the mdx mouse diaphragm. Am. J. Physiol. Cell Physiol. 2011, 300, C1065–C1077. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, S.; Sciorati, C.; D’Antona, G.; Innocenzi, A.; Covarello, D.; Galvez, B.G.; Perrotta, C.; Monopoli, A.; Sanvito, F.; Bottinelli, R.; et al. Nitric oxide release combined with nonsteroidal anti-inflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. USA 2007, 104, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Mammalian models of Duchenne muscular dystrophy: Pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 2011, 184393. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Esaki, K.; Nomura, T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986, 79, 91–95. [Google Scholar] [CrossRef]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of cardiac dystrophy in 3-18-month-old MDX mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.G.; Hahn, H.S.; Wong, B.L.; Lorenz, J.N.; Wenisch, A.S.; Levin, L.S. Evolution of the mdx mouse cardiomyopathy: Physiological and morphological findings. Neuromuscul. Disord. 2004, 14, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Nakamura, K.; Nakao, K.; Kameya, S.; Kobayashi, O.; Nonaka, I.; Kobayashi, T.; Katsuki, M. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 1997, 238, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kameya, S.; Araki, E.; Katsuki, M.; Mizota, A.; Adachi, E.; Nakahara, K.; Nonaka, I.; Sakuragi, S.; Takeda, S.; Nabeshima, Y. Dp260 disrupted mice revealed prolonged implicit time of the b-wave in ERG and loss of accumulation of beta-dystroglycan in the outer plexiform layer of the retina. Hum. Mol. Genet. 1997, 6, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.J.; Winand, N.J.; Stedman, H.; Valentaine, B.A.; Hoffman, E.P.; Kunkel, L.M.; Scott, M.O.; Fishbeck, K.H.; Korneygay, J.N.; Avery, R.J.; et al. The homologue of the Duchenne locus in defective in X-linked muscular dystrophy of dogs. Nature 1988, 334, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Valentine, B.A.; Cooper, B.J.; De Lahunta, R.; O’Quinn, R.; Blue, J.T. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy. Clinical studies. J. Neurol. Sci. 1988, 88, 69–81. [Google Scholar] [CrossRef]
- Shimatsu, Y.; Katagiri, K.; Furuta, T.; Nakura, M.; Tanioka, Y.; Yuasa, K.; Tomohiro, M.; Kornegay, J.N.; Nonaka, I.; Takeda, S. Canine X-linked muscular dystrophy in Japan (CXMDJ). Exp. Anim. 2003, 52, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Sharp, N.J.H.; Kornegay, J.N.; van Camp, S.D.; Herbstreith, M.H.; Secore, S.L.; Kettle, S.; Hung, W.Y.; Constantinou, C.D.; Dykstra, M.J.; Roses, A.D.; et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics 1992, 13, 115–121. [Google Scholar] [CrossRef]
- Shimatsu, Y.; Yoshimura, M.; Yuasa, K.; Urasawa, N.; Tomohiro, M.; Nakura, M.; Tanigawa, M.; Nakamura, A.; Takeda, S. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ. Acta. Myol. 2005, 24, 145–154. [Google Scholar] [PubMed]
- Yugeta, N.; Urasawa, N.; Fujii, Y.; Yoshimura, M.; Yuasa, K.; Wada, M.R.; Nakura, M.; Shimatsu, Y.; Tomohiro, M.; Takahashi, A.; et al. Cardiac involvement in Beagle-based canine X-linked muscular dystrophy in Japan (CXMDJ): Electrocardiographic, echocardiographic, and morphologic studies. BMC Cardiovasc. Disord. 2006, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.L.; Arechavala-Gomeza, V.; Fernandez-Fuente, M.; Burke, M.M.; Nagel, N.; Holder, A.; Stanley, R.; Chandler, K.; Marks, S.L.; Muntoni, F.; et al. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS ONE 2010, 5, e8647. [Google Scholar] [CrossRef] [PubMed]
- Howarth, J.L.; Lee, Y.B.; Uney, J.B. Using viral vectors as gene transfer tools (Cell Biology and Toxicology Special Issue: ETCS-UK 1 day meeting on genetic manipulation of cells). Cell Biol. Toxicol. 2010, 26, 1–20. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.C.; Kobinger, G.P.; Louboutin, J.P.; Radu, A.; Javazon, E.H.; Sena-Esteves, M.; Wilson, J.M.; Flake, A.W. Transduction of satellite cells after prenatal intramuscular administration of lentiviral vectors. J. Gene Med. 2005, 7, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Kafri, T.; Blömer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kimura, E.; Fall, B.M.; Reyes, M.; Angello, J.C.; Welikson, R.; Hauschka, S.D.; Chamberlain, J.S. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene. Ther. 2005, 12, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Counsell, J.R.; Asgarian, Z.; Meng, J.; Ferrer, V.; Vink, C.A.; Howe, S.J.; Waddington, S.N.; Thrasher, A.J.; Muntoni, F.; Morgan, J.E.; et al. Lentiviral vectors can be used for full-length dystrophin gene therapy. Sci. Rep. 2017, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Counsell, J.R.; Asgarian, Z.; Meng, J.; Ferrer, V.; Vink, C.A.; Howe, S.J.; Waddington, S.N.; Thrasher, A.J.; Muntoni, F.; Morgan, J.E.; et al. Lentiviral vectors can be used for full-length dystrophin gene therapy. Sci. Rep. 2017, 7, 44775. [Google Scholar] [CrossRef] [PubMed]
- Rothe, M.; Modlich, U.; Schambach, A. Biosafety challenges for use of lentiviral vectors in gene therapy. Curr. Gene Ther. 2013, 13, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Ragot, T.; Vincent, N.; Chafey, P.; Vigne, E.; Gilgenkrantz, H.; Couton, D.; Cartaud, J.; Briand, P.; Kaplan, J.C.; Perricaudet, M.; et al. Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature 1993, 361, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Jani, A.; Lochmüller, H.; Acsadi, G.; Simoneau, M.; Huard, J.; Garnier, A.; Karpati, G.; Massie, B. Generation, validation, and large scale production of adenoviral recombinants with large size inserts such as a 6.3 kb human dystrophin cDNA. J. Virol. Methods 1997, 64, 111–124. [Google Scholar] [CrossRef]
- Deconinck, N.; Ragot, T.; Maréchal, G.; Perricaudet, M.; Gillis, J.M. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc. Natl. Acad. Sci. USA 1996, 93, 3570–3574. [Google Scholar] [CrossRef] [PubMed]
- Chuah, M.K.; Collen, D.; Vanden Driessche, T. Biosafety of adenoviral vectors. Curr. Gene Ther. 2003, 3, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Yuasa, K.; Yoshimura, M.; Yokota, T.; Ikemoto, T.; Suzuki, M.; Dickson, G.; Miyagoe-Suzuki, Y.; Takeda, S. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002, 293, 1265–1272. [Google Scholar] [CrossRef]
- Yoshimura, M.; Sakamoto, M.; Ikemoto, M.; Mochizuki, Y.; Yuasa, K.; Miyagoe-Suzuki, Y.; Takeda, S. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol. Ther. 2004, 10, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Yoshimura, M.; Urasawa, N.; Ohshima, S.; Howell, J.M.; Nakamura, A.; Hijikata, T.; Miyagoe-Suzuki, Y.; Takeda, S. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 2007, 14, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, S.; Shin, J.H.; Yuasa, K.; Nishiyama, A.; Kira, J.; Okada, T.; Takeda, S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol. Ther. 2009, 17, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Ghosh, A.; Long, C.; Bostick, B.; Smith, B.F.; Kornegay, J.N.; Duan, D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther. 2008, 16, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Chandrasekharan, K.; Xu, R.; Glass, M.; Singhal, N.; Martin, P.T. The synaptic CT carbohydrate modulates binding and expression of extracellular matrix proteins in skeletal muscle: Partial dependence on utrophin. Mol. Cell Neurosci. 2009, 41, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, L.G.; Rodino-Klapac, L.R.; Shao, G.; Xu, R.; Bremer, W.G.; Camboni, M.; Golden, B.; Montgomery, C.L.; Shontz, K.; Heller, K.N.; et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 2014, 22, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Hoshiya, H.; Kazuki, Y.; Abe, S.; Takiguchi, M.; Kajitani, N.; Watanabe, Y.; Yoshino, T.; Shirayoshi, Y.; Higaki, K.; Messina, G.; et al. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol. Ther. 2009, 17, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Uno, N.; Hoshiya, H.; Ragazzi, M.; Ferrari, G.; Kazuki, Y.; Moyle, L.A.; Tonlorenzi, R.; Lombardo, A.; Chaouch, S.; et al. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for Duchenne muscular dystrophy. EMBO Mol. Med. 2018, 10, 254–275. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Shirts, B.H.; Petros, L.M.; Flanigan, K.M.; Gesteland, R.F.; Atkins, J.F. Sequence specificity of aminoglycoside-induced stop codon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann. Neurol. 2000, 48, 164–169. [Google Scholar] [CrossRef]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bönnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- PTC Therapeutics. PTC Therapeutics receives conditional approval in the European Union for Translarna for the treatment of nonsense mutation Duchenne muscular dystrophy. Available online: http://ir.ptcbio.com/releasedetail.cfm?releaseid=863914 (accessed on 27 February 2019).
- Takeshima, Y. Molecular therapies for Duchenne muscular dystrophy. Brain Dev. 2016, 48, 241–246. (In Japanese) [Google Scholar]
- Wilton, S.D.; Dye, D.E.; Blechynden, L.M.; Laing, N.G. Revertant fibres: A possible genetic therapy for Duchenne muscular dystrophy? Neuromuscul. Disord. 1997, 7, 329–335. [Google Scholar] [CrossRef]
- Crawford, G.E.; Lu, Q.L.; Partridge, T.A.; Chamberlain, J.S. Suppression of revertant fibers in mdx mice by expression of a functional dystrophin. Hum. Mol. Genet. 2001, 10, 2745–2750. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem’yeva, O.V.; Strong, P.; Partridge, T.A. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Fletcher, S. Modification of pre-mRNA processing: Application to dystrophin expression. Curr. Opin. Mol. Ther. 2006, 8, 130–135. [Google Scholar] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Antisense-induced multiexon skipping for Duchenne muscular dystrophy makes more sense. Am. J. Hum. Genet. 2004, 74, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.A.; Cau, M.; Muntoni, F.; Mateddu, A.; Galanello, R.; Boccone, L.; Deidda, F.; Loi, D.; Cao, A. Elevation of serum creatine kinase as the only manifestation of an intragenic deletion of the dystrophin gene in three unrelated families. Eur. J. Paediatr. Neurol. 1998, 2, 255–261. [Google Scholar] [CrossRef]
- Schwartz, M.; Duno, M.; Palle, A.L.; Krag, T.; Vissing, J. Deletion of exon 16 of the dystrophin gene is not associated with disease. Hum. Mutat. 2007, 28, 205. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Pistilli, E.; Duddy, W.; Nagaraju, K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2007, 7, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J. Hum. Genet. 2017, 62, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Exon-skipping therapy for Duchnne muscular dystrophy. Neuropathology 2009, 29, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Mouly, V.; Garcia, L.; Yokota, T.; Duddy, W. In silico screening based on predictive algorithms as a design tool for exon skipping of oligonucleotides in Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0120058. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Wilton, S.D. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 2006, 8, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscle of mdx mice. Mol. Ther. 2008, 16, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Veltrop, M.; van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; van der Kaa, J.; Linssen, M.M.; den Dunnen, J.T.; Verbeek, S.; et al. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Carroll, J. Muscular Dystrophy Drug from GlaxoSmithKline, Prosensa Fails PhIII. FierceBiotechol. 20 September 2013. Available online: http://www.fiercebiotech.com/story/muscular-dystrophy-drug-glaxosmithkline-prosensa-fails-phiii/2013-09-20 (accessed on 27 February 2019).
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.C.; Bhasin, N.; Tewari, M.; Brown, A.E.; Safer, D.; Sweeney, H.L.; Discher, D.E. Exon-skipped dystrophins for treatment of Duchenne muscular dystrophy: Mass spectrometry mapping of most exons and cooperative domain designs based on single molecule mechanics. Cytoskeleton 2010, 67, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S. Approves Controversial FDA. DMD drug amid scientific controversy and advocates’ pleas. Neurol. Today 2016, 16, 8–12. [Google Scholar]
- Mendell, J.R.; Goemans, N.; Lowes, L.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group and Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O.H.; Finkel, R.S.; Rummey, C.; Benton, M.J.; Glanzman, A.M.; Flickinger, J.; Lindström, B.M.; Meier, T. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr. Pulmonol. 2015, 50, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Science Trans. Med. 2018, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nagata, T.; Yokota, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. High efficient in vivo delivery of PMO in to regenerating myotubes and rescue in laminin-α2 chain null congenital muscular dystrophy mice. Hum. Mol. Genet. 2013, 22, 4914–4928. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoshida, K.; Fukushima, K.; Ueda, H.; Urasawa, N.; Koyama, J.; Yazaki, Y.; Yazaki, M.; Sakai, T.; Haruta, S.; et al. Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 2008, 15, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Taglia, A.; Petillo, R.; D’Ambrosio, P.; Picillo, E.; Torella, A.; Orsini, C.; Ergoli, M.; Scutifero, M.; Passamano, L.; Palladino, A.; et al. Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 2015, 34, 9–13. [Google Scholar] [PubMed]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S. Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. Crispr-mediated genome editing restores dystrophin expression and function in MDX mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. Crispr/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight. 2017, 2, 18. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| DNA Analysis | Mutational Type (Frequency) | Note | |
|---|---|---|---|
| MLPA | Deletions of one or more exons (60–70%) Duplications (5–10%) | Mutational type: DMD: out-of-frame mutation BMD: in-frame mutation | |
| Direct sequencing | Single nucleotide variants Small deletions or insertions Single-base changes Splice site changes | (20–35% in total) | Frequency: DMD: 25–35% BMD: 10–20% |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, A. Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy. J. Pers. Med. 2019, 9, 16. https://doi.org/10.3390/jpm9010016
Nakamura A. Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy. Journal of Personalized Medicine. 2019; 9(1):16. https://doi.org/10.3390/jpm9010016
Chicago/Turabian StyleNakamura, Akinori. 2019. "Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy" Journal of Personalized Medicine 9, no. 1: 16. https://doi.org/10.3390/jpm9010016
APA StyleNakamura, A. (2019). Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy. Journal of Personalized Medicine, 9(1), 16. https://doi.org/10.3390/jpm9010016