Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy



Abstract
1. Introduction
2. Theory of CRISPR Application for Gene Editing
3. DMD Studies In Vitro and Alternatives to Viral Delivery of CRISPR
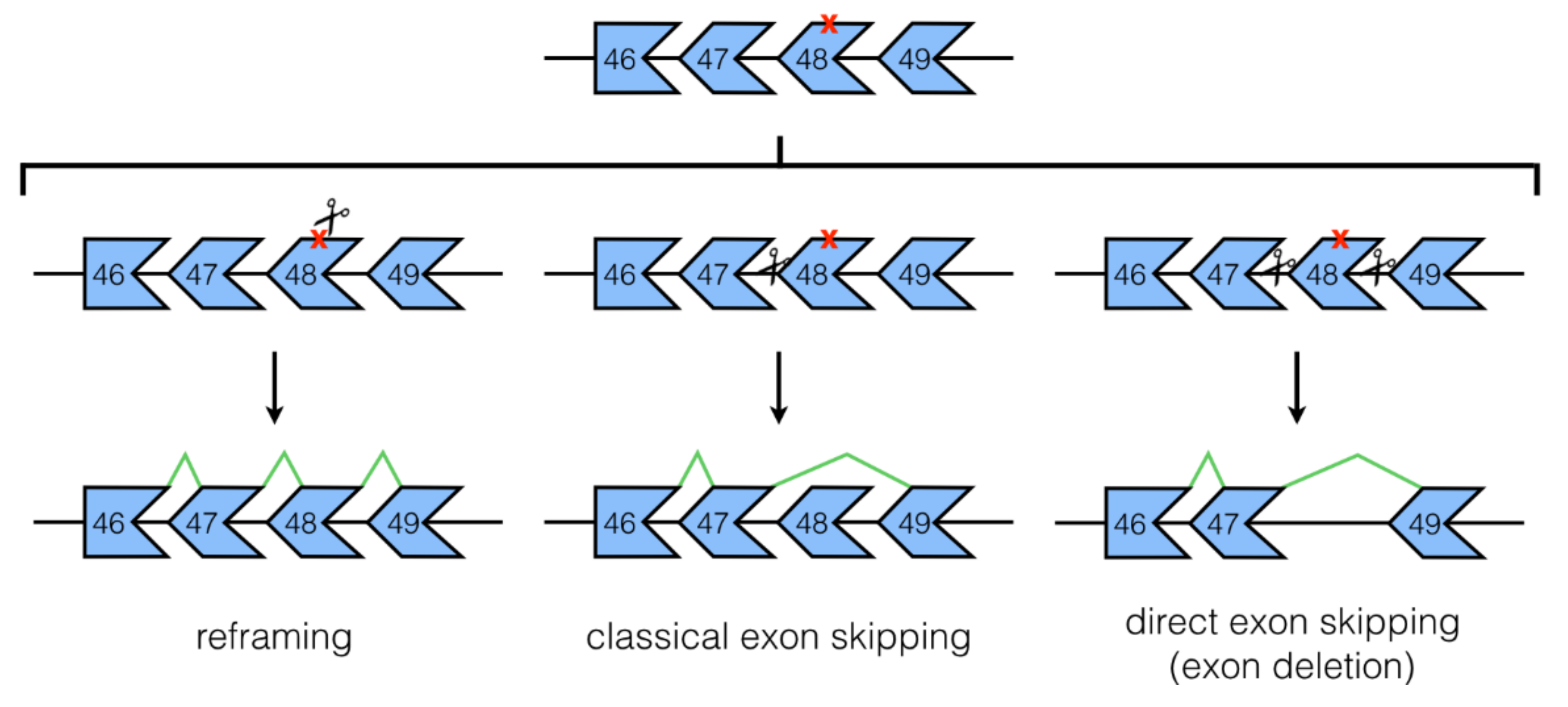
3.1. NHEJ Mechanisms: Classical and Direct Exon Skipping
3.2. Base Editing
3.3. Utrophin Upregulation
4. DMD Studies In Vivo
4.1. Studies in DMD Mouse Models
4.2. Study in a DMD Dog Model
4.3. CRISPR/Cas9 for the Generation of DMD Animal/Cell Models
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- O’Brien, K.F.; Kunkel, L.M. Dystrophin and muscular dystrophy: Past, present, and future. Mol. Genet. Metab. 2001, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Landfeldt, E.; Sejersen, T.; Tulinius, M. A mini review and implementation model for using ataluren to treat nonsense mutation duchenne muscular dystrophy. Acta Paediatr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Tyagi, R.; Mohanty, M.; Goyal, M.; Silva, K.R.; Wijekoon, N. Dystrophin induced cognitive impairment: Mechanisms, models and therapeutic strategies. Ann. Neurosci. 2015, 22, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K. Current and emerging treatment strategies for duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016, 12, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.G.; Coffey, A.J.; Bobrow, M.; Bentley, D.R. Exon structure of the human dystrophin gene. Genomics 1993, 16, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Abbs, S.; Tuffery-Giraud, S.; Bakker, E.; Ferlini, A.; Sejersen, T.; Mueller, C.R. Best practice guidelines on molecular diagnostics in duchenne/becker muscular dystrophies. Neuromuscul. Disord. 2010, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Aslesh, T.; Maruyama, R.; Yokota, T. Skipping multiple exons to treat DMD-promises and challenges. Biomedicines 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Yokota, T. Invention and early history of exon skipping and splice modulation. Methods Mol. Biol. 2018, 1828, 3–30. [Google Scholar] [PubMed]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, Cpf1 and C2C1/2/3-What’s next? Bioengineered 2017, 8, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, L.; He, X.; Liu, X.; Zhou, C.; Liu, J.; Ge, X.; Li, J.; Liu, C.; Zhao, J.; et al. SaCas9 requires 5′-NNGRRT-3′ PAM for sufficient cleavage and possesses higher cleavage activity than SpCas9 or FnCpf1 in human cells. Biotechnol. J. 2018, 13, e1800080. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M. Studying cardiac arrhythmias in the mouse—A reasonable model for probing mechanisms? Trends Cardiovasc. Med. 2004, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Salama, G.; London, B. Mouse models of long QT syndrome. J. Physiol. 2007, 578, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed]
- Price, P.S.; Keenan, R.E.; Swartout, J.C. Characterizing interspecies uncertainty using data from studies of anti-neoplastic agents in animals and humans. Toxicol. Appl. Pharmacol. 2008, 233, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wei, G.; Li, P.; Zhou, X.; Zhang, Y. Urine-derived stem cells: A novel and versatile progenitor source for cell-based therapy and regenerative medicine. Genes Dis. 2014, 1, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The treat-nmd DMD global database: Analysis of more than 7000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by talen and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Wojtal, D.; Kemaladewi, D.U.; Malam, Z.; Abdullah, S.; Wong, T.W.; Hyatt, E.; Baghestani, Z.; Pereira, S.; Stavropoulos, J.; Mouly, V.; et al. Spell checking nature: Versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet. 2016, 98, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Iyombe-Engembe, J.P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lague, P.; Tremblay, J.P. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the cindel method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Goncalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. Crispr-mediated genome editing restores dystrophin expression and function in MDX mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Goncalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, A.; Duguez, S.; Moiani, A.; Izmiryan, A.; Barbon, E.; Martin, S.; Mamchaoui, K.; Mouly, V.; Bernardi, F.; Mavilio, F.; et al. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol. Ther. Nucleic Acids 2017, 7, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. Crispr/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Mokhonova, E.; Quinonez, M.; Pyle, A.D.; Spencer, M.J. Creation of a novel humanized dystrophic mouse model of duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J. Neuromuscul. Dis. 2017, 4, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. Crispr/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Lu-Nguyen, N.B.; Malerba, A.; Kim, E.; Kim, D.; Cappellari, O.; Cho, H.Y.; Dickson, G.; Popplewell, L.; Kim, J.S. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using campylobacter jejuni Cas9. Mol. Ther. 2018, 26, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Duchene, B.L.; Cherif, K.; Iyombe-Engembe, J.P.; Guyon, A.; Rousseau, J.; Ouellet, D.L.; Barbeau, X.; Lague, P.; Tremblay, J.P. CRISPR-induced deletion with saCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol. Ther. 2018, 26, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell 2018, 72, 380–394.e7. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Wood, M.J. Development of multiexon skipping antisense oligonucleotide therapy for duchenne muscular dystrophy. Biomed. Res. Int. 2013, 2013, 402369. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tome, F.M.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991, 1, 185–194. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Ervasti, J.M.; Matsumura, K.; Kahl, S.D.; Leveille, C.J.; Campbell, K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron 1991, 7, 499–508. [Google Scholar] [CrossRef]
- Clerk, A.; Morris, G.E.; Dubowitz, V.; Davies, K.E.; Sewry, C.A. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J. 1993, 25, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Jasmin, B.J. Utrophin upregulation for treating duchenne or becker muscular dystrophy: How close are we? Trends Mol. Med. 2006, 12, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, M.; Negri, T.; Cozzi, F.; Colpo, R.; Andreetta, F.; Croci, D.; Davies, K.E.; Cornelio, F.; Pozza, O.; Karpati, G.; et al. Dystrophic phenotype of canine x-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003, 10, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Tinsley, J.M.; Phelps, S.R.; Squire, S.E.; Townsend, E.R.; Martin, J.E.; Davies, K.E. Non-toxic ubiquitous over-expression of utrophin in the MDX mouse. Neuromuscul. Disord. 2001, 11, 713–721. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- t Hoen, P.A.; de Meijer, E.J.; Boer, J.M.; Vossen, R.H.; Turk, R.; Maatman, R.G.; Davies, K.E.; van Ommen, G.J.; van Deutekom, J.C.; den Dunnen, J.T. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem. 2008, 283, 5899–5907. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Ryu, S.-M.; Kim, S.-T.; Baek, G.; Kim, D.; Lim, K.; Chung, E.; Kim, S.; Kim, J.-S. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol. 2017, 35, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.L.; Arechavala-Gomeza, V.; Fernandez-Fuente, M.; Burke, M.M.; Nagel, N.; Holder, A.; Stanley, R.; Chandler, K.; Marks, S.L.; Muntoni, F.; et al. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS ONE 2010, 5, e8647. [Google Scholar] [CrossRef] [PubMed]
- Hildyard, J.C.W.; Taylor-Brown, F.; Massey, C.; Wells, D.J.; Piercy, R.J. Determination of qpcr reference genes suitable for normalizing gene expression in a canine model of duchenne muscular dystrophy. J. Neuromuscul. Dis. 2018, 5, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/cas system. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.H.; Zhao, H.; Qing, Y.B.; Pan, W.R.; Jia, B.Y.; Zhao, H.Y.; Huang, X.X.; Wei, H.J. Porcine zygote injection with Cas9/sgRNA results in DMD-modified pig with muscle dystrophy. Int. J. Mol. Sci. 2016, 17, 1668. [Google Scholar] [CrossRef] [PubMed]
- Sui, T.; Lau, Y.S.; Liu, D.; Liu, T.; Xu, L.; Gao, Y.; Lai, L.; Li, Z.; Han, R. A novel rabbit model of duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 2018, 11, dmm032201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Hosoki, K.; Nakatsuji, Y.; Yokota, T.; Obika, S. A novel human muscle cell model of duchenne muscular dystrophy created by CRISPR/Cas9 and evaluation of antisense-mediated exon skipping. J. Hum. Genet. 2018, 63, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Yokota, T. Creation of DMD muscle cell model using CRISPR-Cas9 genome editing to test the efficacy of antisense-mediated exon skipping. Methods Mol. Biol. 2018, 1828, 165–171. [Google Scholar] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Crispr off-targets: A reassessment. Nat. Methods 2018, 15, 229–230. [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. Crispr off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.E.; Gillum, D.; Kiani, S. Approaches to reduce CRISPR off-target effects for safer genome editing. Appl. Biosaf. 2017, 22, 7–13. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered pam specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. Crispr–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1408. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.-M.; Akyüz, L.; Reinke, P.; Volk, H.-D.; Schmueck-Henneresse, M. High prevalence of streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.E.; Deeconda, A.; Tsang, S.H. Viral vectors, engineered cells and the CRISPR revolution. Adv. Exp. Med. Biol. 2017, 1016, 3–27. [Google Scholar] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cas9 Enzyme | Source Bacteria | PAM Site | Protein, Gene Size | Key Features | Reference(s) |
|---|---|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | 5′-NGG-3′ | 1368 aa, 4.10 kbp | Ubiquitous PAM site, widely used with multiples derivatives | [27,28,29] |
| SaCas9 | Staphylococcus aureus | 5′-NNGRRT-3′ | 1053 aa, 3.16 kbp | Smaller size, better packaged for viral delivery | [30,31] |
| CjCas9 | Campylobacter jejuni | 5′-NNNNACAC-3′, 5′-NNNNRYAC-3′ | 984 aa, 2.95 kbp | Even smaller than SaCas9, lower chance of off-targeting due to longer PAM | [32] |
| Cas Enzyme | Strategy | Target Gene Region(s) | Model(s) | Delivery | Study Highlights | Reference |
|---|---|---|---|---|---|---|
| SpCas9 | NHEJ reframing, HDR exon correction | Dmd exon 23 | mdx mice | 1-cell embryo injection | Dystrophin restoration observed by IHC (up to 100%) and WB; 17% Dmd HDR correction resulted in 47–60% dystrophin-positive fibers in skeletal muscles and the heart | 2014 Long et al. [43] |
| SpCas9 | NHEJ reframing, exon skipping, HDR exon knock-in | DMD intron 44/exon 45 | DMD hiPSCs, hiPSC-derived skeletal muscle cells (ex44 del.) | Electroporation | Dystrophin restoration in derived skeletal muscle cells observed by WB and IHC for all strategies; CRISPR was as effective as using TALEN | 2015 Li et al. [41] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 45–55 (for reframing each exon), introns 50 and 51 (ex51 del.), introns 44 and 55 (ex45–55 del.) | immortalized DMD patient muscle cells (ex48–50 del.), immunodeficient NSG mice | Electroporation | Generated targeted deletions of exon/s in vitro, particularly of the large exon 45–55 region which led to dystrophin rescue by WB; mice transplanted with treated myoblasts (exon 51-deleted) showed dystrophin-positive fibers by IHC | 2015 Ousterout et al. [21] |
| dSpCas9-VP16 | Utrophin upregulation | UTRN A/B promoter | immortalized DMD patient muscle cells (ex45–52 del.) | Electroporation | 1.7–6.9-fold upregulation of utrophin achieved; restored β-dystroglycan expression observed by WB with as little as 1.7-fold upregulation | 2016 Wojtal et al. [44] |
| SpCas9 | Duplicated exons removal | DMD intron 27 | primary DMD patient fibroblasts (ex18–30 dup.) | LV transduction, with Adeno-MyoD | 4.42% full-length dystrophin production achieved post-treatment, accompanied with α-dystroglycan restoration | 2016 Wojtal et al. [44] |
| SpCas9 | Single exon deletion | Dmd exon 23, introns 22 and 23 (ex23 del.) | mdx mice | AAV9 delivery (i.m., i.p., i.v.) | All modes of injection led to appearance of dystrophin-positive fibers as evaluated by IHC: ~25.5% 6 wks post-i.m., ~4.6% and ~9.6% in skeletal and cardiac muscles respectively 12 wks post-i.v., ~1.8% and ~3.2% in skeletal and cardiac muscles respectively 8 wks post-i.p. | 2016 Long et al. [45] |
| SaCas9 | Single exon deletion | Dmd introns 22 and 23 (ex23 del.) | mdx mice | AAV8 delivery (i.m., i.p., i.v.) | Intramuscular injections led to ~59% of transcripts with exon 23 deleted, which restored about 8% dystrophin of healthy levels by WB, proper relocalization of DGC proteins, and muscle function improvement; systemic injections restored dystrophin production in the heart and skeletal muscles | 2016 Nelson et al. [46] |
| SpCas9, SaCas9 | Single exon deletion | Dmd introns 22 and 23 (ex23 del.) | mdx mice, mdx satellite cells | AAV9 delivery (i.m., i.p., i.v.) | Dual-vector (Cas9 and gRNAs on separate constructs) had higher cutting efficiency than a single-vector system (Cas9 and gRNAs on the same construct) in vitro; dystrophin restoration >10% observed in the heart and skeletal muscles upon systemic treatment; correction also possible in satellite cells | 2016 Tabebordbar et al. [47] |
| SpCas9 | Hybrid exon formation via internal exon deletion | DMD exons 50 and 54 | immortalized DMD patient muscle cells (ex51–53 del.), hDMD/mdx mice | Lipotransfection (in vitro)/ electroporation (in vivo) | Dystrophin restoration successful in vitro by WB, not shown in vivo; hybrid exon formation thought to preserve dystrophin rod domain structure better | 2016 Iyombe-Engembe et al. [48] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 51, 53, introns 52 and 53 (ex53 del.), 43 and 54 (ex44–54 del.) | immortalized DMD patient muscle cells (ex48–50, or 45–52 del.) | Sequential LV then AdV transduction/AdV transduction | Study showed the possibility of combining both TALEN and CRISPR approaches in one gene editing strategy; also, comparable editing was obtained with Cas9 and gRNA delivered either together or separately in AdV | 2016 Maggio et al. [49] |
| SpCas9 | Multiple exon deletion | Dmd introns 20 and 23 (ex21–23 del.) | mdx mice | Electroporation/AdV transduction | Treatment restored proper calcium dynamics in muscle (electroporation), and restored dystrophin to 50% of wild-type levels, as well as dystrophin-associated complex sarcolemmal localization and muscle membrane integrity (transduction) | 2016 Xu et al. [50] |
| SpCas9 | Multiple exon deletion | DMD introns 44 and 55 (ex45–55 del.) | DMD hiPSCs, hiPSC-derived skeletal and cardiac muscle cells (ex46–51 or 46–47 del., ex50 dup.), immunodeficient NSG-mdx mice | Nucleofection | CRISPR-mediated deletion of the large exon 45–55 region achieved, restored membrane function and dystrophin, β-dystroglycan expression by WB and IHC; mice transplanted with hiPSC-derived skeletal muscle cells showed dystrophin-positive fibers by IHC | 2016 Young et al. [42] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 51, 53 (for reframing) introns 52 and 53 (ex53 del.), introns 43 and 54 (ex44–54 del.) | immortalized DMD patient muscle cells (ex48–50, or 45–52 del.) | AdV transduction | AdV with 2gRNA-SpCas9 constructs work as good as those with 1gRNA-SpCas9 constructs in terms of corrective ability and dystrophin restoration | 2016 Maggio et al. [51] |
| SpCas9, SaCas9 | Multiple exon deletion, HDR exon correction | Dmd exon 53, introns 51 and 53 (ex52–53 del.) | mdx4cv mice (nonsense ex53 mutation) | AAV6 delivery (i.m., i.v.) | Dual vector approach (SpCas9 and gRNA separate) yielded higher correction efficiency than single vector approach (SaCas9 and gRNA together); systemic treatment restored dystrophin expression in the heart (~34% dystrophin-positive fibers) and skeletal muscles (~10–50% dystrophin-positive fibers) | 2017 Bengtsson et al. [52] |
| LbCpf1, AsCpf1 | NHEJ reframing, single exon skipping, HDR exon correction | DMD exon 51, intron 50 | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex48–50 del.), mdx mice | Nucleofection (in vitro)/ 1-cell embryo injection (in vivo) | Cpf1 editing successfully restored dystrophin expression and improved mitochondrial function in cardiomyocytes; 5/24 pups (injected at the embryo stage) showed HDR correction and had ameliorated dystrophic phenotypes | 2017 Zhang et al. [53] |
| SpCas9 | Duplicated exon removal | DMD exon 2, intron 2 | immortalized DMD patient muscle cells (ex2 dup.) | PEI transfection/LV transduction | Use of a single gRNA can delete a duplicated exon, resulting in slight dystrophin rescue by WB and IHC | 2017 Lattanzi et al. [54] |
| SpCas9 | HDR exon correction | Dmd exon 23 | mdx mice, mdx satellite cells | Lipotransfection (template, gRNA), AdV transduction (Cas9)/AdV transduction | Higher transduction efficiency obtained when AdVs were used for both Cas9 and gRNA-HDR template delivery; mice transplanted with corrected satellite cells showed dystrophin-positive fibers by IHC | 2017 Zhu et al. [55] |
| SpCas9 | Multiple exon deletion | DMD introns 44 and 55 (ex45–55 del.) | humanized mdx mice with DMD exon 45 del. | Electroporation | Exon 45–55 deletion by CRISPR possible in vivo; first use of the humanized DMD mouse model with exon 45 del. for CRISPR studies | 2017 Young et al. [56] |
| SpCas9 | Multiple exon deletion | DMD introns 2 and 7 (ex3–9 del.), introns 5 and 7 (ex6–7 del.), introns 6 and 11 (ex7–11 del.) | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex8–9 or ex3–7 del.) | Nucleofection | Dystrophin with ex7–11 del. showed the least functionality, while those with ex3–9 del. had the highest functionality in terms of assessing iPSC-derived cardiomyocyte calcium cycling | 2017 Kyrychenko et al. [57] |
| SpCas9 | HDR correction | Dmd exon 23 | mdx primary muscle cells, mdx mice | CRISPR-Gold nanoparticles (i.m.) | 5.4% HDR correction of the Dmd mutation in mdx was observed after CRISPR treatment and cardiotoxin injection, dystrophin-positive fibers found by IHC; 0.8% HDR correction observed without cardiotoxin co-injection, which led to significantly improved hanging test performance | 2017 Lee et al. [58] |
| SpCas9 | NHEJ reframing, single exon skipping | Dmd exon 51 | mice with Dmd exon 50 del. | AAV9 delivery (i.m., i.p.) | Successful dystrophin restoration in the heart and skeletal muscles; systemic injections led to improved muscle function; first application of CRISPR in the ex50 del. mouse model | 2017 Amoasii et al. [59] |
| SpCas9 | Single exon deletion | Dmd introns 50 and 51 (ex51 del.) | primary human skeletal muscle cells | HCAdV delivery | Up to 93.3% exon 51 deletion observed in vitro upon delivery of CRISPR agents by HCAdV | 2017 Ehrke-Schulz et al. [60] |
| SpCas9 | NHEJ reframing, exon skipping | DMD exon 51, introns 47, 50, 54 | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex48–50 del., pseudo-ex47, ex55–59 dup.) | Nucleofection | All strategies corrected the respective patient mutations and restored dystrophin production in iPSC-derived cardiomyocytes; 3D-engineered heart muscle produced from treated iPSC-derived cardiomyocytes showed improved contractile force | 2018 Long et al. [61] |
| CjCas9 | NHEJ reframing | Dmd exon 23 | mice with deletions in Dmd exon 23 | AAV9 delivery (i.m.) | CjCas9 displayed higher targeting specificity than SpCas9; use of CjCas9-based CRISPR can lead to successful dystrophin restoration and improvement in muscle function as well | 2018 Koo et al. [62] |
| SaCas9 | Hybrid exon formation via multiple exon deletion | DMD exons 47 and 58 | DMD skeletal muscle cells (ex51–53 del., ex49–50 del., ex51–56 del., ex50–52 del.), humanized mdx mice with DMD ex52 del. | LV transduction (in vitro)/AAV9 delivery (in vivo; i.v.) | gRNAs designed to produce exon deletions that best preserved dystrophin protein structure were able to show dystrophin restoration in vitro and in vivo (slight rescue in the heart) | 2018 Duchêne et al. [63] |
| SpCas9 | NHEJ reframing, exon skipping | Dystrophin exon 51 | deltaE50-MD canine model (ex50 del.) | AAV9 delivery (i.m., i.v.) | First published study on dystrophin gene correction in a dog model; ~3–70% dystrophin restoration of healthy levels in skeletal muscles and ~92% in the heart found by WB | 2018 Amoasii et al. [64] |
| nSpCas9-ABE7.10 | Base editing to correct a nonsense mutation | Dmd exon 20 | mice with a nonsense mutation in Dmd exon 20 | trans-splicing AAV2/9 delivery (i.m.) | ~3.3% base editing frequency achieved 8 weeks post-treatment with no detectable off-target effects; ~17% dystrophin-positive fibers and restored localization of nNOS observed by IHC | 2018 Ryu et al. [65] |
| dSa/SpCas9-TAM | Base editing to induce exon skipping | DMD intron 50 5′ splice site | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex51 del.) | Lipotransfection | ~100% base editing efficiency achieved; corrected iPSC-derived cardiomyocytes had restored dystrophin protein, low CK and miR-31 levels, and restoration of β-dystroglycan expression | 2018 Yuan et al. [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.R.Q.; Yoon, C.; Yokota, T. Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. J. Pers. Med. 2018, 8, 38. https://doi.org/10.3390/jpm8040038
Lim KRQ, Yoon C, Yokota T. Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. Journal of Personalized Medicine. 2018; 8(4):38. https://doi.org/10.3390/jpm8040038
Chicago/Turabian StyleLim, Kenji Rowel Q., Chantal Yoon, and Toshifumi Yokota. 2018. "Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy" Journal of Personalized Medicine 8, no. 4: 38. https://doi.org/10.3390/jpm8040038
APA StyleLim, K. R. Q., Yoon, C., & Yokota, T. (2018). Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. Journal of Personalized Medicine, 8(4), 38. https://doi.org/10.3390/jpm8040038