
AI-Guided Chemotherapy Optimization in Lung Cancer Using Genomic and Survival Data

Abstract
1. Introduction
2. Materials and Methods
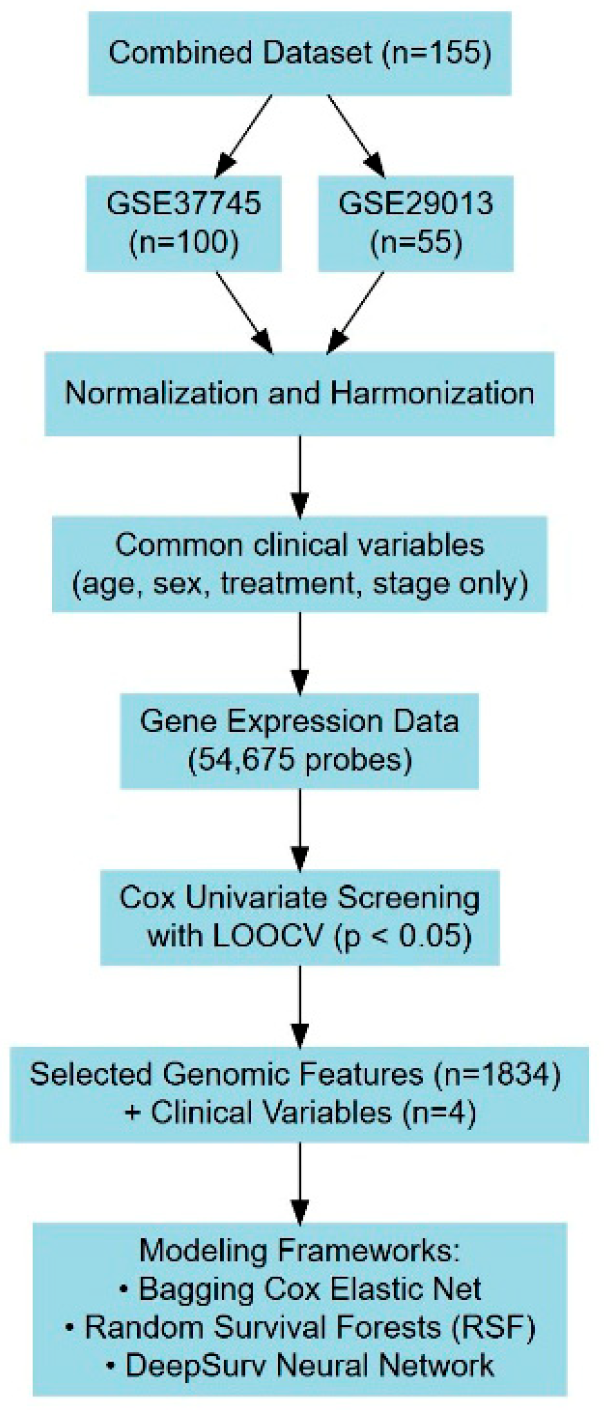
2.1. Data Description
2.2. Model Design
2.2.1. Regularized Cox Proportional Hazards Model
2.2.2. Random Survival Forests Model
2.2.3. Deep Learning Survival Model
2.2.4. Performance Measure: Concordance Index
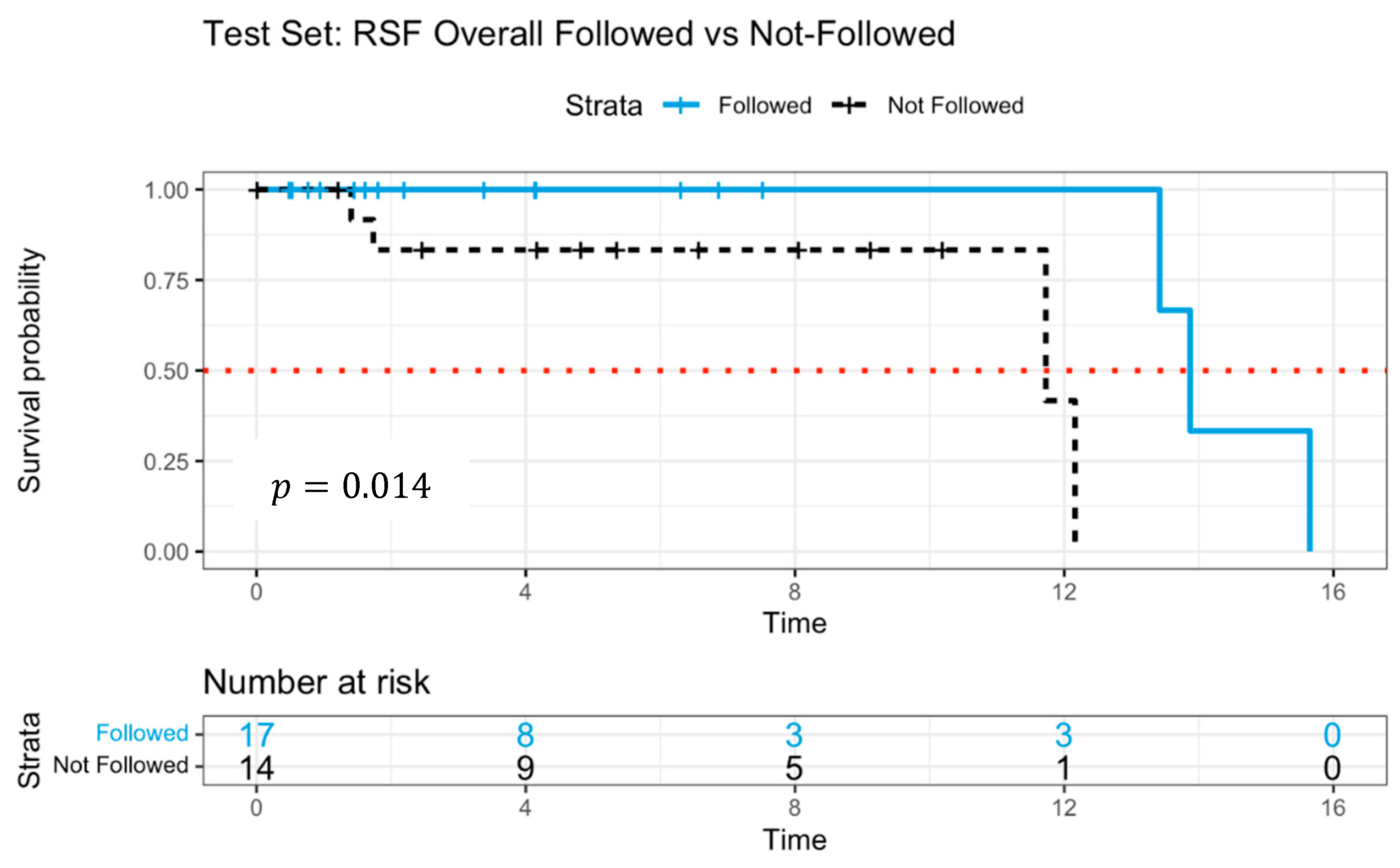
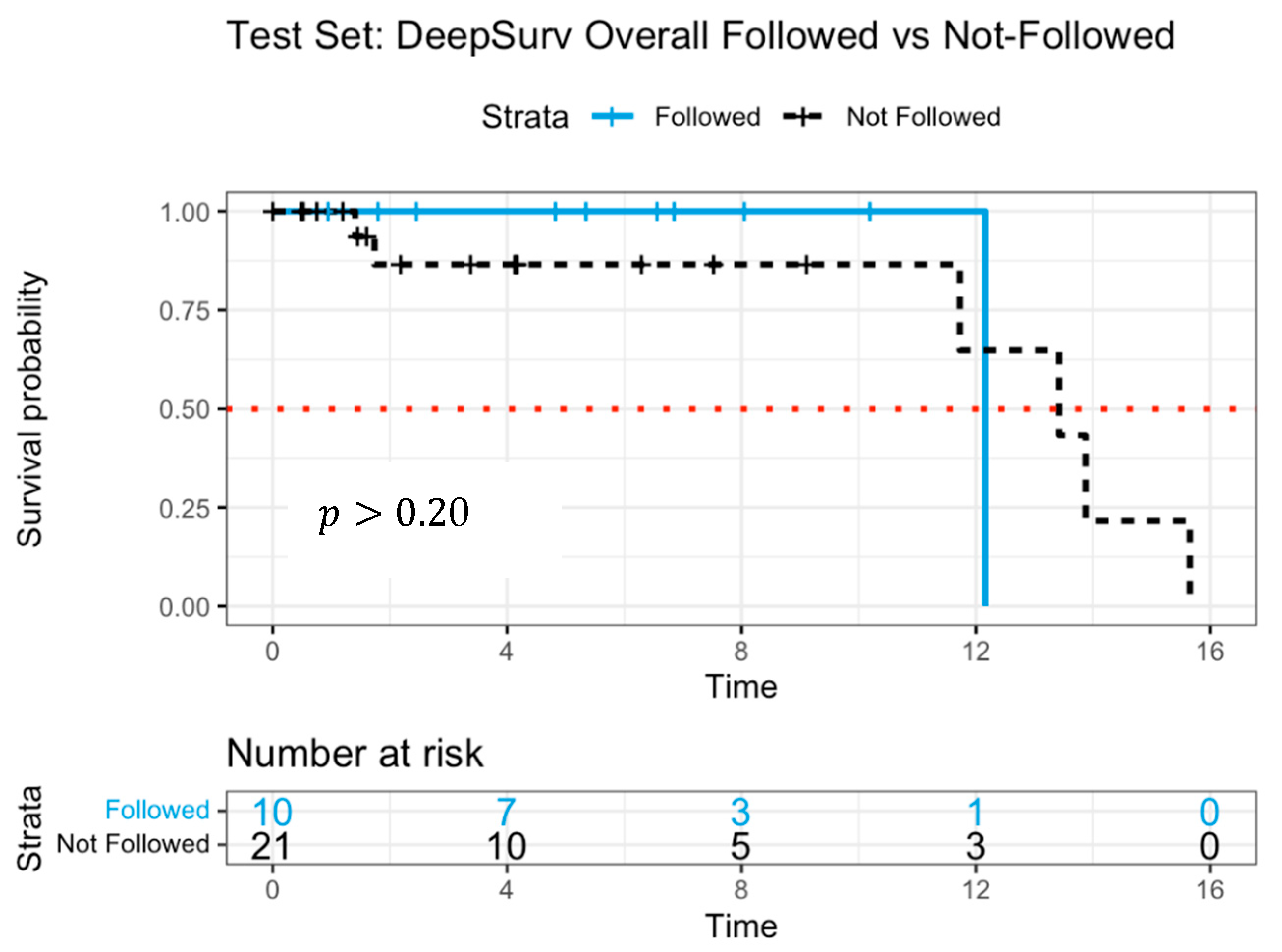
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef]
- Pirker, R.; Filipits, M. Adjuvant Therapy in Patients With Completely Resected Non-Small-Cell Lung Cancer: Current Status and Perspectives. Clin. Lung Cancer 2019, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine Learning Applications in Cancer Prognosis and Prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef]
- Cruz, J.A.; Wishart, D.S. Applications of Machine Learning in Cancer Prediction and Prognosis. Cancer Inform. 2006, 2, 59–77. [Google Scholar] [CrossRef]
- Miotto, R.; Wang, F.; Wang, S.; Jiang, X.; Dudley, J.T. Deep Learning for Healthcare: Review, Opportunities and Challenges. Brief. Bioinform. 2018, 19, 1236–1246. [Google Scholar] [CrossRef]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.S.; Thrun, S.; Dean, J. A Guide to Deep Learning in Healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Hosny, A.; Schabath, M.B.; Giger, M.L.; Birkbak, N.J.; Mehrtash, A.; Allison, T.; Arnaout, O.; Abbosh, C.; Dunn, I.F.; et al. Artificial Intelligence in Cancer Imaging: Clinical Challenges and Applications. CA Cancer J. Clin. 2019, 69, 127–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Clayton, E.A.; Matyunina, L.V.; McDonald, L.D.; Benigno, B.B.; McDonald, J.F. Machine Learning Predicts Individual Cancer Patient Responses to Therapeutic Drugs with High Accuracy. Sci. Rep. 2021, 11, 12110. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J. High-Performance Medicine: The Convergence of Human and Artificial Intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]
- Moon, H.; Zhao, Y.; Pluta, D.; Ahn, H. Subgroup Analysis Based on Prognostic and Predictive Gene Signatures for Adjuvant Chemotherapy in Early-Stage Non-Small-Cell Lung Cancer Patients. J. Biopharm. Stat. 2018, 28, 750–762. [Google Scholar] [CrossRef]
- Moon, H.; Chao, T.; Ahn, H. Identification of Risk Factors and Likelihood of Benefit from Adjuvant Chemotherapy for Early-Stage Lung Cancer Patients. J. Biopharm. Stat. 2020, 30, 430–444. [Google Scholar] [CrossRef]
- Moon, H.; Nguyen, A.; Lee, E. Prognostic Genomic Predictive Biomarkers for Early-Stage Lung Cancer Patients. Open Biomark. J. 2021, 11, 69–78. [Google Scholar] [CrossRef]
- Moon, H.; Tran, L.; Lee, A.; Kwon, T.; Lee, M. Prediction of Treatment Recommendations Via Ensemble Machine Learning Algorithms for Non-Small Cell Lung Cancer Patients in Personalized Medicine. Cancer Inform. 2024, 23, 11769351241272397. [Google Scholar] [CrossRef]
- Breiman, L. Bagging Predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Ishwaran, H.; Kogalur, U.B.; Blackstone, E.H.; Lauer, M.S. Random Survival Forests. Ann. Appl. Stat. 2008, 2, 841–860. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized Treatment Recommender System Using a Cox Proportional Hazards Deep Neural Network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Botling, J.; Edlund, K.; Lohr, M.; Hellwig, B.; Holmberg, L.; Lambe, M.; Berglund, A.; Ekman, S.; Bergqvist, M.; Pontén, F.; et al. Biomarker Discovery in Non-Small Cell Lung Cancer: Integrating Gene Expression Profiling, Meta-Analysis, and Tissue Microarray Validation. Clin. Cancer Res. 2013, 19, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xiao, G.; Coombes, K.R.; Behrens, C.; Solis, L.M.; Raso, G.; Girard, L.; Erickson, H.S.; Roth, J.; Heymach, J.V.; et al. Robust Gene Expression Signature from Formalin-Fixed Paraffin-Embedded Samples Predicts Prognosis of Non-Small-Cell Lung Cancer Patients. Clin. Cancer Res. 2011, 17, 5705–5714. [Google Scholar] [CrossRef]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B Methodol. 1972, 34, 187–202. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar]
- Ishwaran, H.; Kogalur, U.B. Fast Unified Random Forests for Survival, Regression, and Classification (RF-SRC); R Package Version 3.3.3. 2025. Available online: https://cran.r-project.org/package=randomForestSRC (accessed on 20 August 2024).
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical Bayesian Optimization of Machine Learning Algorithms. Adv. Neural Inf. Process. Syst. 2012, 25, 2951–2959. [Google Scholar] [CrossRef]
- Bjorck, J.; Gomes, C.; Selman, B.; Weinberger, K. Understanding Batch Normalization. In Proceedings of the 32nd International Conference on Neural Information Processing Systems (NeurIPS 2018), Montreal, ON, Canada, 2–8 December 2018; Curran Associates Inc.: Red Hook, NY, USA, 2018; pp. 7705–7716. Available online: https://dl.acm.org/doi/pdf/10.5555/3327757.3327868 (accessed on 12 October 2024).
- Klambauer, G.; Unterthiner, T.; Mayr, A.; Hochreiter, S. Self-Normalizing Neural Networks. In Proceedings of the Advances in Neural Information Processing Systems 30 (NeurIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Curran Associates Inc.: Long Beach, CA, USA, 2017; pp. 971–980. Available online: https://proceedings.neurips.cc/paper/2017/file/5d44ee6f2c3f71b73125876103c8f6c4-Paper.pdf (accessed on 12 October 2024).
- Harrell, F.E., Jr.; Califf, R.M.; Pryor, D.B.; Lee, K.L.; Rosati, R.A. Evaluating the Yield of Medical Tests. JAMA 1982, 247, 2543–2546. [Google Scholar] [CrossRef]
- Uno, H.; Cai, T.; Pencina, M.J.; D’Agostino, R.B.; Wei, L.J. On the C-Statistic for Evaluating Overall Adequacy of Risk Prediction Procedures with Censored Survival Data. Stat. Med. 2011, 30, 1105–1117. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr.; Lee, K.L.; Mark, D.B. Multivariable Prognostic Models: Issues in Developing Models, Evaluating Assumptions and Adequacy, and Measuring and Reducing Errors. Stat. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]
- Pencina, M.J.; D’Agostino, R.B. Overall C as a Measure of Discrimination in Survival Analysis: Model-Specific Population Value and Confidence Interval Estimation. Stat. Med. 2004, 23, 2109–2123. [Google Scholar] [CrossRef]
- Steyerberg, E.W.; Van Calster, B.; Pencina, M.J. Performance Measures for Prediction Models and Their Evaluation in the Presence of Censoring. Stat. Methods Med. Res. 2018, 27, 2504–2525. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Amer. Statist. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- SynGO Consortium. SynGO—ID Conversion Tool. SynGO.org. 2019. Available online: https://www.syngoportal.org/convert (accessed on 12 October 2024).
- Shimura, T.; Shibata, M.; Inoue, T.; Owada-Ozaki, Y.; Yamaura, T.; Muto, S.; Hasegawa, T.; Shio, Y.; Suzuki, H. Prognostic Impact of Serum Transthyretin in Patients with Non-Small Cell Lung Cancer. Mol. Clin. Oncol. 2019, 10, 597–604. [Google Scholar] [CrossRef]
- Sedo, A.; Krepela, E.; Kasafírek, E. Dipeptidyl Peptidase IV, Prolyl Endopeptidase and Cathepsin B Activities in Primary Human Lung Tumors and Lung Parenchyma. J. Cancer Res. Clin. Oncol. 1991, 117, 249–253. [Google Scholar] [CrossRef]
- Liu, L.; Song, X.; Li, X.; Xue, L.; Ding, S.; Niu, L.; Xie, L.; Song, X. A Three-Platelet mRNA Set: MAX, MTURN and HLA-B as Biomarker for Lung Cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2713–2723. [Google Scholar] [CrossRef]
- Enfield, K.S.S.; Marshall, E.A.; Anderson, C.; Ng, K.W.; Rahmati, S.; Xu, Z.; Fuller, M.; Milne, K.; Lu, D.; Shi, R.; et al. Epithelial Tumor Suppressor ELF3 Is a Lineage-Specific Amplified Oncogene in Lung Adenocarcinoma. Nat. Commun. 2019, 10, 5438. [Google Scholar] [CrossRef] [PubMed]
- Ay, A.S.; Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 Constitutes a Native Store-Operated Calcium Entry That Regulates Non-Small Cell Lung Adenocarcinoma Cell Proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef]
- Kim, D.S.; Kwon, N.S.; Yun, H.Y. Leucine Rich Repeat LGI Family Member 3: Integrative Analyses Reveal Its Prognostic Association with Non-Small Cell Lung Cancer. Oncol. Lett. 2019, 18, 3388–3398. [Google Scholar] [CrossRef]
- Wang, X.W.; Guo, Q.Q.; Wei, Y.; Ren, K.M.; Zheng, F.S.; Tang, J.; Zhang, H.Y.; Zhao, J.G. Construction of a Competing Endogenous RNA Network Using Differentially Expressed lncRNAs, miRNAs and mRNAs in Non-Small Cell Lung Cancer. Oncol. Rep. 2019, 42, 2402–2415. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.B.; Xu, C.; Wang, W.; Zhang, Y.Z.; Huang, J.M.; Xie, Y.J.; Wang, Q.Q.; Fan, X.X.; Yao, X.J.; Xie, C.; et al. Plumbagin Suppresses Non-Small Cell Lung Cancer Progression through Downregulating ARF1 and by Elevating CD8+ T Cells. Pharmacol. Res. 2021, 169, 105656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhao, L.; Yuan, H.; Xu, L.; Tan, W.; Song, Y.; Fang, X. A New Small Cell Lung Cancer Biomarker Identified by Cell-SELEX Generated Aptamers. Exp. Cell Res. 2019, 382, 111478. [Google Scholar] [CrossRef]
- Ren, W.; Yue, C.; Liu, L.; Du, L.; Xu, K.; Zhou, Y. Overexpression of Bruton Tyrosine Kinase Inhibits the Proliferation, Migration, and Invasion of Non-Small Cell Lung Cancer Cells. Anal. Cell Pathol. 2023, 2023, 3377316. [Google Scholar] [CrossRef]
- Xu, F.; Gu, J.; Lu, C.; Mao, W.; Wang, L.; Zhu, Q.; Liu, Z.; Chu, Y.; Liu, R.; Ge, D. Calpain-2 Enhances Non-Small Cell Lung Cancer Progression and Chemoresistance to Paclitaxel via EGFR-pAKT Pathway. Int. J. Biol. Sci. 2019, 15, 127–137. [Google Scholar] [CrossRef]
- Chen, Q.; Shen, L.; Li, S. Emerging Role of Inositol Monophosphatase in Cancer. Biomed. Pharmacother. 2023, 161, 114442. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gu, J.; Kuang, X.; Yu, Y.; Rao, B.; Fang, S.; Lu, J.; Qiu, F. An Integrative Pan-Cancer Analysis of WWC Family Genes and Functional Validation in Lung Cancer. Cell Signal. 2024, 115, 111034. [Google Scholar] [CrossRef]
- Zhu, J.; Mou, Y.; Ye, S.; Hu, H.; Wang, R.; Yang, Q.; Hu, Y. Identification of a Six-Gene SLC Family Signature with Prognostic Value in Patients with Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 803198. [Google Scholar] [CrossRef]
- Harjes, U. E-selectin fills two needs for metastasis. Nat. Rev. Cancer. 2019, 19, 301. [Google Scholar] [CrossRef]
- Sansregret, L.; Patterson, J.O.; Dewhurst, S.; López-García, C.; Koch, A.; McGranahan, N.; Chao, W.C.H.; Barry, D.J.; Rowan, A.; Instrell, R.; et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017, 7, 218–233. [Google Scholar] [CrossRef]
- He, C.; Huang, D.; Yang, F.; Huang, D.; Cao, Y.; Peng, J.; Luo, X. High Expression of lncRNA HEIH Is Helpful in the Diagnosis of Non-Small Cell Lung Cancer and Predicts Poor Prognosis. Cancer Manag. Res. 2022, 14, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Cascone, R.; Carlucci, A.; Pierdiluca, M.; Santini, M.; Fiorelli, A. Prognostic Value of Soluble Major Histocompatibility Complex Class I Polypeptide-Related Sequence A in Non-Small-Cell Lung Cancer—Significance and Development. Lung Cancer 2017, 8, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Gu, J.; Lu, C.; Lin, J.; Stewart, D.; Chang, D.; Roth, J.A.; Wu, X. Genetic Variations in the Regulator of G-Protein Signaling Genes Are Associated with Survival in Late-Stage Non-Small Cell Lung Cancer. PLoS ONE 2011, 6, e21120. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Yan, X.; Diao, Y.; Cui, D.; Liu, H. T Cell Differentiation Protein 2 Facilitates Cell Proliferation by Enhancing mTOR-Mediated Ribosome Biogenesis in Non-Small Cell Lung Cancer. Discov. Oncol. 2022, 13, 26. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Li, L.; Qin, Y.R.; Liu, H.; Jiang, C.; Zeng, T.T.; Li, M.Q.; Xie, D.; Li, Y.; et al. TSPAN15 Interacts with BTRC to Promote Oesophageal Squamous Cell Carcinoma Metastasis via Activating NF-κB Signaling. Nat. Commun. 2018, 9, 1423. [Google Scholar] [CrossRef]
- Jen, J.; Wang, Y.C. Zinc Finger Proteins in Cancer Progression. J. Biomed. Sci. 2016, 23, 53. [Google Scholar] [CrossRef]
- Takata, Y.; Xiang, Y.B.; Burk, R.F.; Li, H.; Hill, K.E.; Cai, H.; Gao, J.; Zheng, W.; Shu, X.O.; Cai, Q. Plasma Selenoprotein P Concentration and Lung Cancer Risk: Results from a Case-Control Study Nested within the Shanghai Men’s Health Study. Carcinogenesis 2018, 39, 1352–1358. [Google Scholar] [CrossRef]
- Li, P.; Liu, H.; Zhang, Z.; Lv, X.; Wang, H.; Ma, J.; Ma, Z.; Qu, X.; Teng, Y.E. Expression and Comparison of Cbl-b in Lung Squamous Cell Carcinoma and Adenocarcinoma. Med. Sci. Monit. 2018, 24, 623–635. [Google Scholar] [CrossRef]
- Yen, M.C.; Wu, K.L.; Liu, Y.W.; Chang, Y.Y.; Chang, C.Y.; Hung, J.Y.; Tsai, Y.M.; Hsu, Y.L. Ubiquitin Conjugating Enzyme E2 H (UBE2H) Is Linked to Poor Outcomes and Metastasis in Lung Adenocarcinoma. Biology 2021, 10, 378. [Google Scholar] [CrossRef]
- Zou, Z.; Zhang, B.; Li, Z.; Lei, L.; Sun, G.; Jiang, X.; Guan, J.; Zhang, Y.; Xu, S.; Li, Q. KBTBD7 Promotes Non-Small Cell Lung Carcinoma Progression by Enhancing Ubiquitin-Dependent Degradation of PTEN. Cancer Med. 2022, 11, 4544–4554. [Google Scholar] [CrossRef]
- Cui, Y.; Song, Y.; Yan, S.; Cao, M.; Huang, J.; Jia, D.; Liu, Y.; Zhang, S.; Fan, W.; Cai, L.; et al. CUEDC1 Inhibits Epithelial-Mesenchymal Transition via the TβRI/Smad Signaling Pathway and Suppresses Tumor Progression in Non-Small Cell Lung Cancer. Aging 2020, 12, 20047–20068. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.K.; Kundu, S.T.; Bajaj, R.; Rodriguez, B.L.; Fradette, J.J.; Gibbons, D.L. MBIP (MAP3K12 Binding Inhibitory Protein) Drives NSCLC Metastasis by JNK-Dependent Activation of MMPs. Oncogene 2020, 39, 6719–6732. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, R.; Guo, Y.; Loehrer, E.; Wei, Y.; Zhu, Y.; Yuan, Q.; Moran, S.; Fleischer, T.; Bjaanaes, M.M.; et al. A Multi-Omic Study Reveals BTG2 as a Reliable Prognostic Marker for Early-Stage Non-Small Cell Lung Cancer. Mol. Oncol. 2018, 12, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Majumder, M.; Mackiewicz, K.; Howley, B.V.; Peterson, Y.K.; Howe, P.H.; Palanisamy, V.; Diehl, J.A. Fbxo4-Mediated Degradation of Fxr1 Suppresses Tumorigenesis in Head and Neck Squamous Cell Carcinoma. Nat. Commun. 2017, 8, 1534. [Google Scholar] [CrossRef]
- El-Khoury, V.; Béland, M.; Schritz, A.; Kim, S.Y.; Nazarov, P.V.; Gaboury, L.; Sertamo, K.; Bernardin, F.; Batutu, R.; Antunes, L.; et al. Identification of Beta-Arrestin-1 as a Diagnostic Biomarker in Lung Cancer. Br. J. Cancer 2018, 119, 580–590. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, X.; Xu, Z.; He, Y.; Guo, C.; He, L.; Huan, C.; Cai, C.; Huang, J.; Zhang, J.; et al. PDLIM5 Inhibits STUB1-Mediated Degradation of SMAD3 and Promotes the Migration and Invasion of Lung Cancer Cells. J. Biol. Chem. 2020, 295, 13798–13811. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set (n = 124) | Testing Set (n = 31) | |
|---|---|---|
| Treatment Received | ||
| Adjuvant Chemotherapy (ACT) | 50 | 13 |
| Observation (OBS) | 74 | 18 |
| Age | ||
| Less than 65 | 49 | 17 |
| Older than or equal to 65 | 75 | 14 |
| Stage of Disease | ||
| I | 74 | 15 |
| II | 25 | 8 |
| III | 24 | 8 |
| IV | 1 | 0 |
| Variable | Variable Importance Score | Gene Symbol | Gene Descriptions from SynGo Consortium [38] |
|---|---|---|---|
| 209660_at | 0.008556 | TTR | Transthyretin [39] |
| 212215_at | 0.007373 | PREPL | prolyl endopeptidase like [40] |
| 227000_at | 0.007349 | MTURN | maturin, neural progenitor differentiation regulator homolog [41] |
| 227200_at | 0.006915 | ETV3 | ETS variant transcription factor 3 [42] |
| 218811_at | 0.006703 | ORAI2 | ORAI calcium release-activated calcium modulator 2 [43] |
| 228886_at | 0.006037 | LRRC27 | leucine-rich repeat containing 27 [44] |
| 240184_at | 0.006008 | SYNPR-AS1 | SYNPR antisense RNA 1 [45] |
| 218230_at | 0.005832 | ARFIP1 | ADP ribosylation factor interacting protein 1 [46] |
| 225012_at | 0.005657 | HDLBP | high-density lipoprotein binding protein [47] |
| 205504_at | 0.005625 | BTK | Bruton tyrosine kinase [48] |
| 208683_at | 0.005619 | CAPN2 | calpain 2 [49] |
| 203126_at | 0.005215 | IMPA2 | inositol monophosphatase 2 [50] |
| 225273_at | 0.005053 | WWC3 | WWC family member 3 [51] |
| 207249_s_at | 0.004782 | SLC28A2 | solute carrier family 28 member 2 [52] |
| 206211_at | 0.004512 | SELE | selectin E [53] |
| 229145_at | 0.004417 | ANAPC16 | anaphase promoting complex subunit 16 [54] |
| 226146_at | 0.004407 | HEIH | hepatocellular carcinoma up-regulated EZH2-associated long non-coding RNA [55] |
| 235352_at | 0.004392 | MR1 | major histocompatibility complex, class I-related [56] |
| 234297_at | 0.004382 | RGS8 and SDHAP3 | regulator of G protein signaling 8 and SDHA pseudogene 3 [57] |
| 224650_at | 0.004321 | MAL2 | mal, T cell differentiation protein 2 [58] |
| 218693_at | 0.004226 | TSPAN15 | tetraspanin 15 [59] |
| 218707_at | 0.004064 | ZNF444 | zinc finger protein 444 [60] |
| 233167_at | 0.003896 | SELENOO | selenoprotein O [61] |
| 209682_at | 0.003893 | CBLB | Cbl proto-oncogene B [62] |
| 200667_at | 0.003872 | UBE2D3 | ubiquitin-conjugating enzyme E2 D3 [63] |
| 229970_at | 0.003856 | KBTBD7 | kelch repeat and BTB domain containing 7 [64] |
| 219468_s_at | 0.003791 | CUEDC1 | CUE domain containing 1 [65] |
| 205448_s_at | 0.003735 | MAP3K12 | mitogen-activated protein kinase kinase kinase 12 [66] |
| 201236_s_at | 0.003712 | BTG2 | BTG anti-proliferation factor 2 [67] |
| 214623_at | 0.003702 | FBXW4P1 | F-box and WD repeat domain containing 4 pseudogene 1 [68] |
| 221861_at | 0.003697 | ARRB1 | arrestin beta 1 [69] |
| 241208_at | 0.003691 | PDLIM5 | PDZ and LIM domain 5 [70] |
| Model | Assumptions | Handles Nonlinearity/Interactions | Training C-Index | Test C-Index | Survival Curve Separation | Interpretability | Notable Strengths |
|---|---|---|---|---|---|---|---|
| Bagging Cox (Elastic Net) | Proportional hazards, linear effects | Limited (via penalization only) | 0.996 | 0.709 | Moderate | High | Simple, interpretable, stable with bagging |
| Random Survival Forest (RSF) | Nonparametric | Yes | 0.889 | 0.885 | Strong | Moderate | Best test performance, good at capturing interactions |
| DeepSurv Neural Network | Flexible, neural Cox model | Yes (deep architecture) | 0.990 | 0.982 | Weak to moderate | Low | High predictive accuracy, handles complex relationships |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, H.; Nguyen, P.N.; Park, J.; Lee, M.; Ahn, S. AI-Guided Chemotherapy Optimization in Lung Cancer Using Genomic and Survival Data. J. Pers. Med. 2025, 15, 218. https://doi.org/10.3390/jpm15060218
Moon H, Nguyen PN, Park J, Lee M, Ahn S. AI-Guided Chemotherapy Optimization in Lung Cancer Using Genomic and Survival Data. Journal of Personalized Medicine. 2025; 15(6):218. https://doi.org/10.3390/jpm15060218
Chicago/Turabian StyleMoon, Hojin, Phan N. Nguyen, Jaehee Park, Minho Lee, and Sohyul Ahn. 2025. "AI-Guided Chemotherapy Optimization in Lung Cancer Using Genomic and Survival Data" Journal of Personalized Medicine 15, no. 6: 218. https://doi.org/10.3390/jpm15060218
APA StyleMoon, H., Nguyen, P. N., Park, J., Lee, M., & Ahn, S. (2025). AI-Guided Chemotherapy Optimization in Lung Cancer Using Genomic and Survival Data. Journal of Personalized Medicine, 15(6), 218. https://doi.org/10.3390/jpm15060218