

Discovery and Validation of a SIT1-Related Prognostic Signature Associated with Immune Infiltration in Cutaneous Melanoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Correlation between SIT1 and Tumor Immune Cell Infiltration
2.3. Immunomodulators
2.4. Construction of the Prognostic Model
2.5. Construction of Nomogram
2.6. Statistical Analysis
3. Results
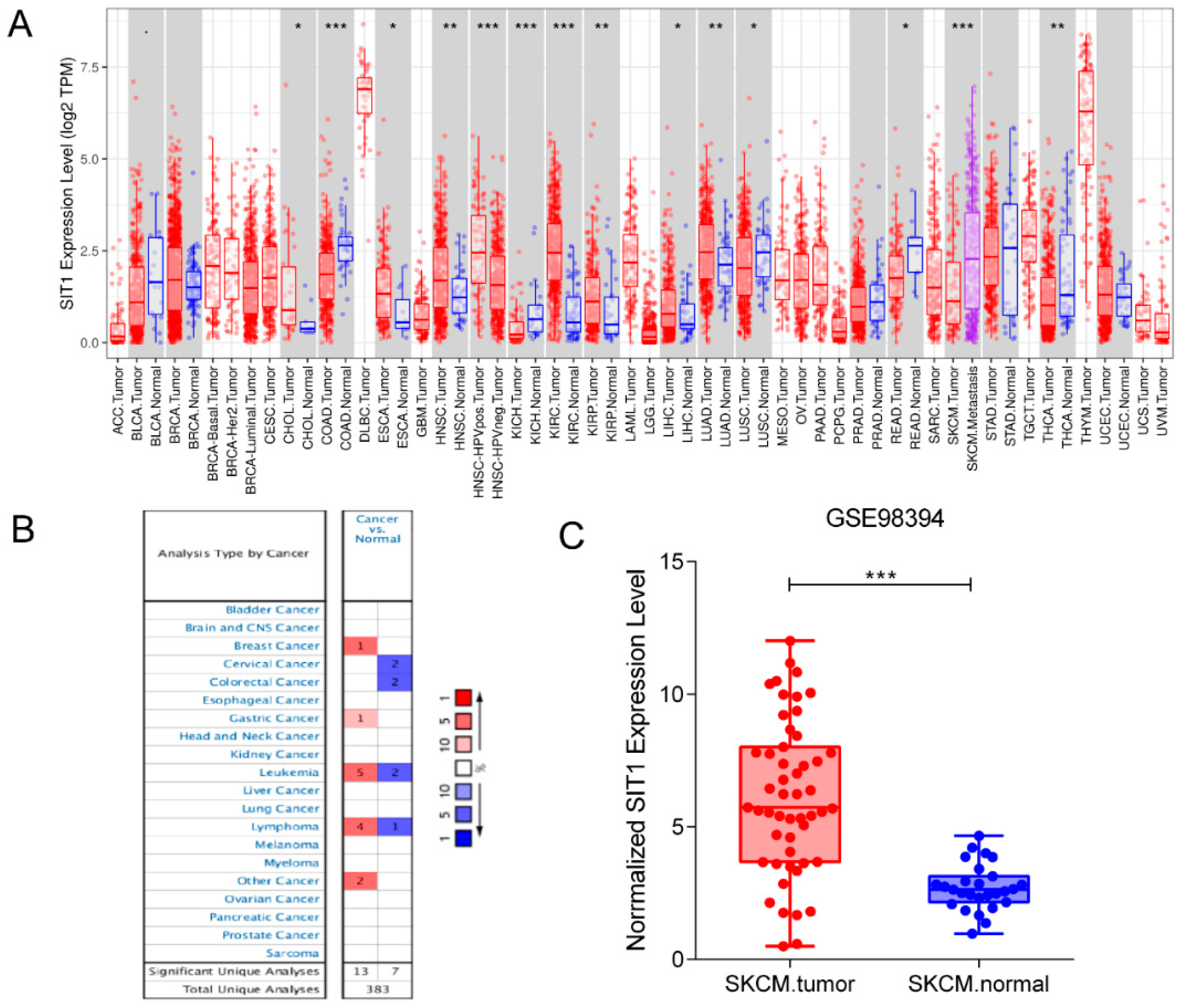
3.1. mRNA Level of SIT1 in Various Tumor Types
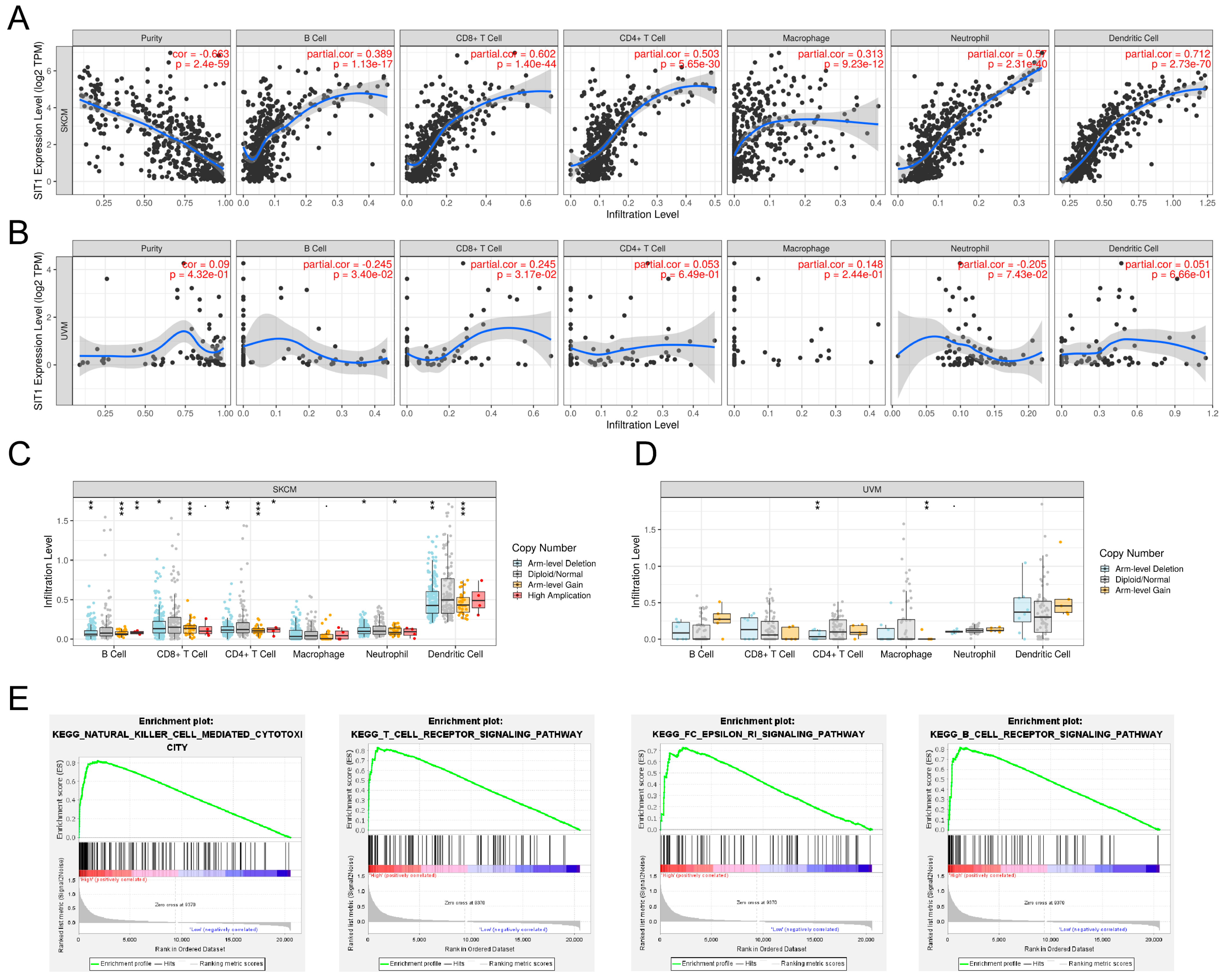
3.2. Association between SIT1 and Immune Cells
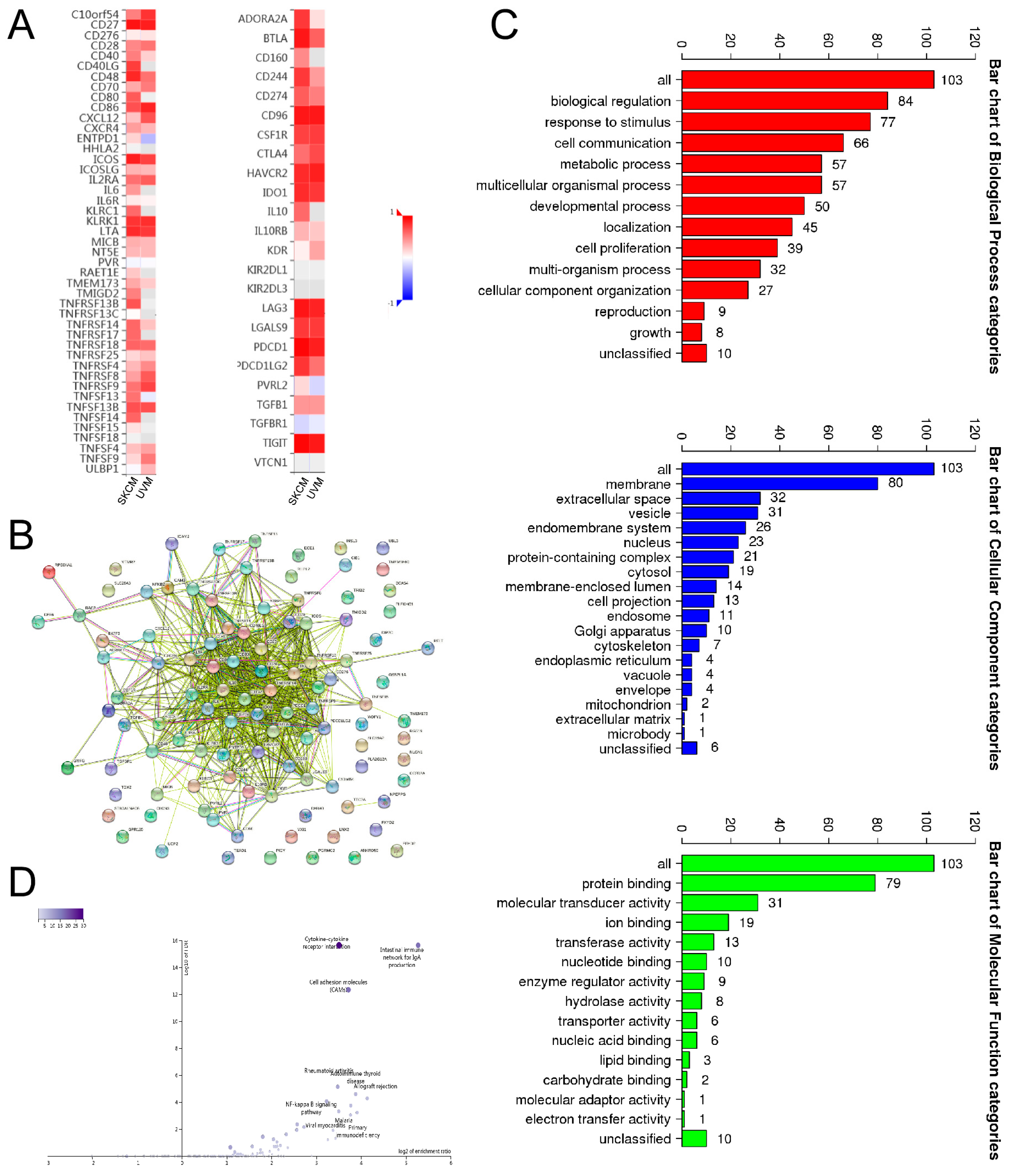
3.3. Association between SIT1 Expression and Immune Markers
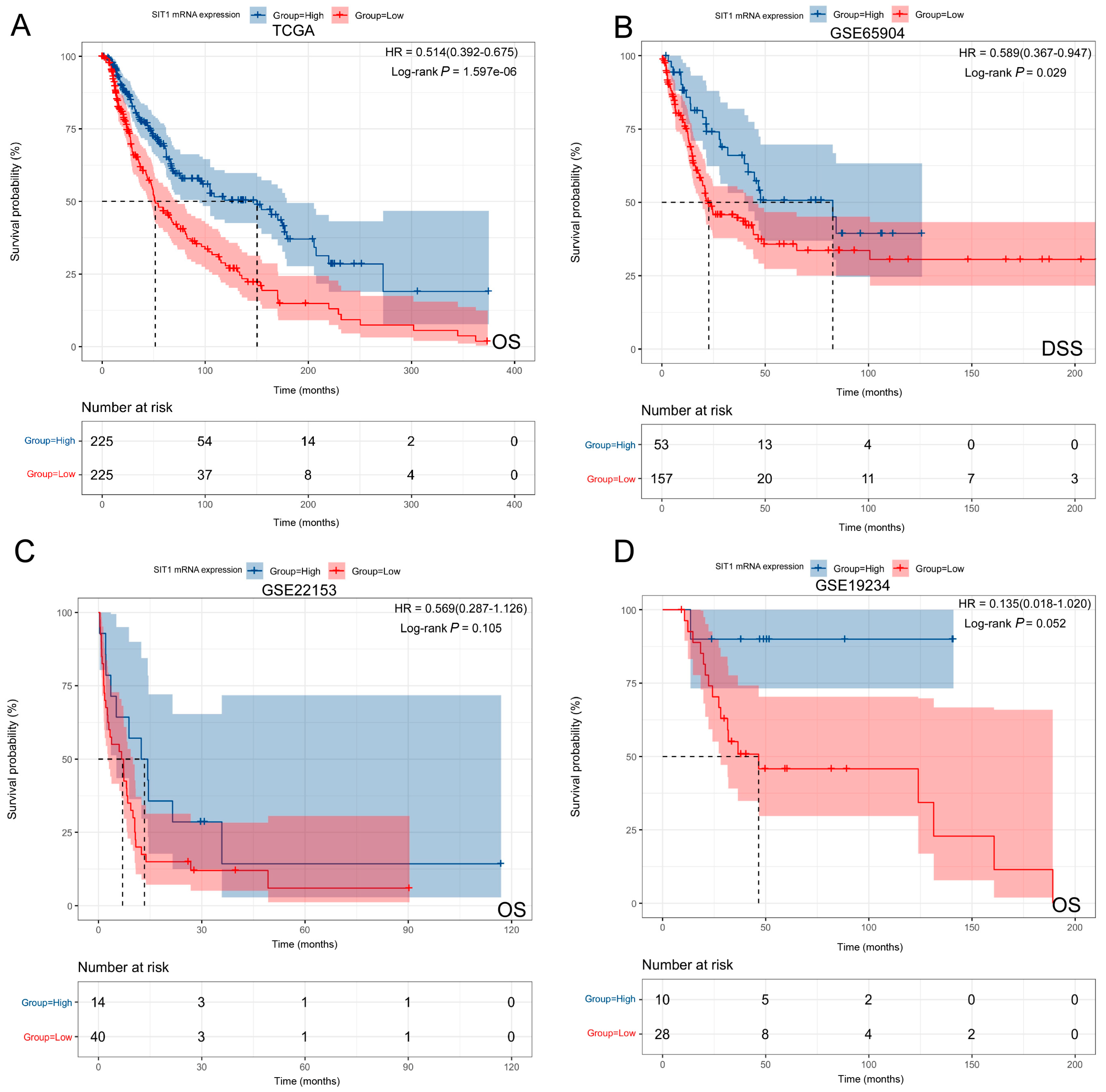
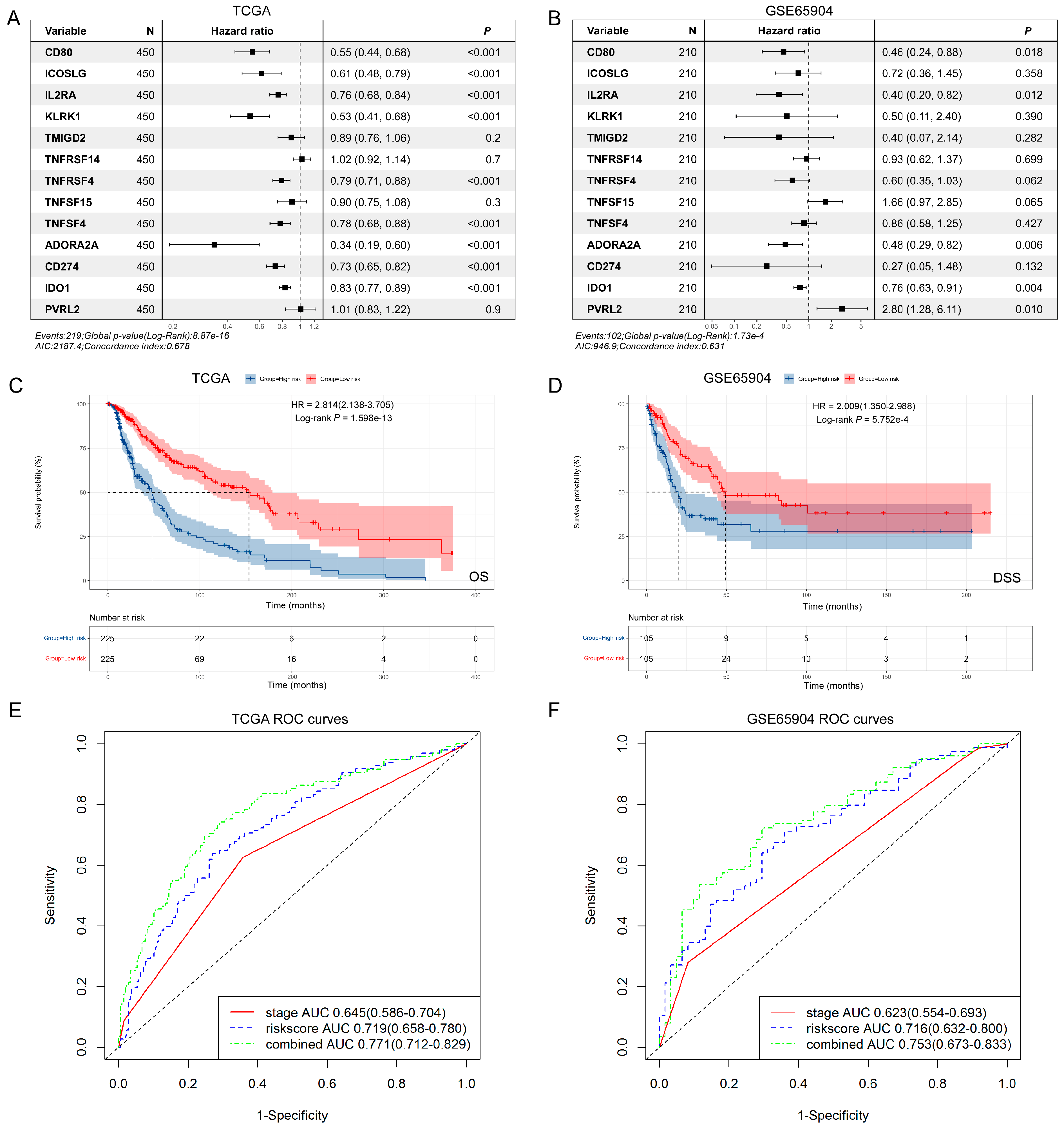
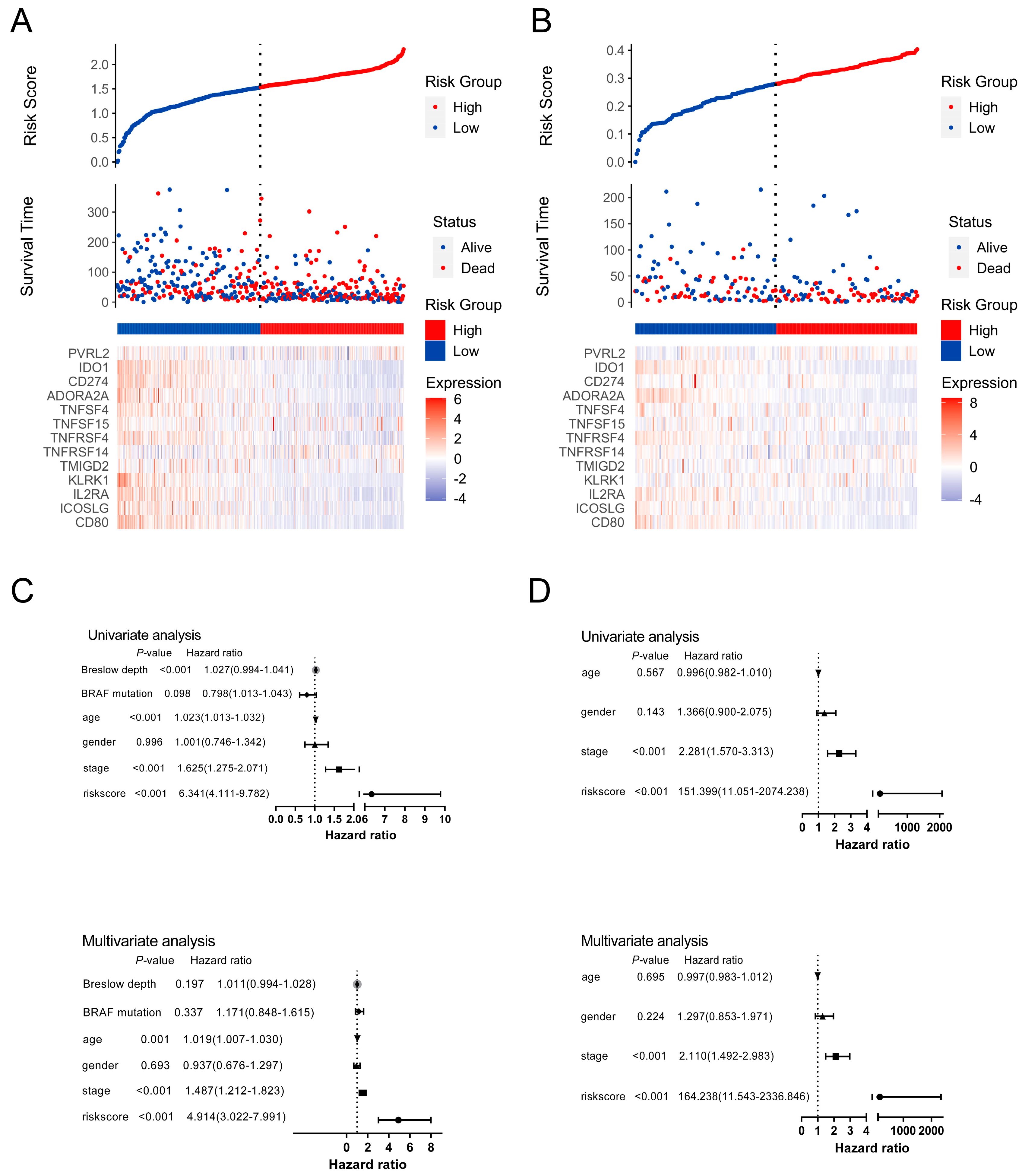
3.4. The Prognostic Implication of SIT1 and SIT1-Associated Immunomodulators in SKCM
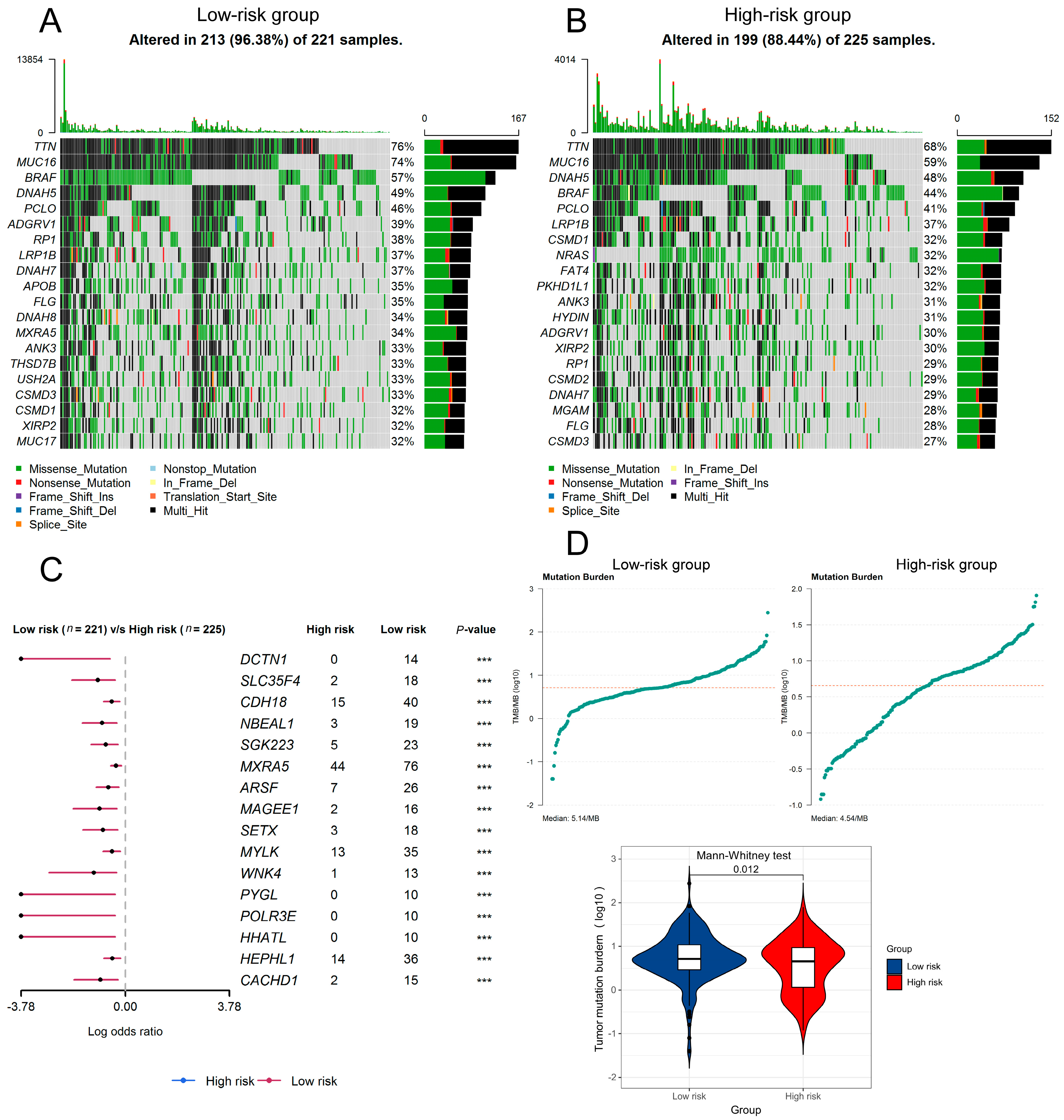
3.5. The Predictive Value of SIT1-Associated Immunomodulators Signature for the Efficacy of Immunotherapy on Melanoma Patients
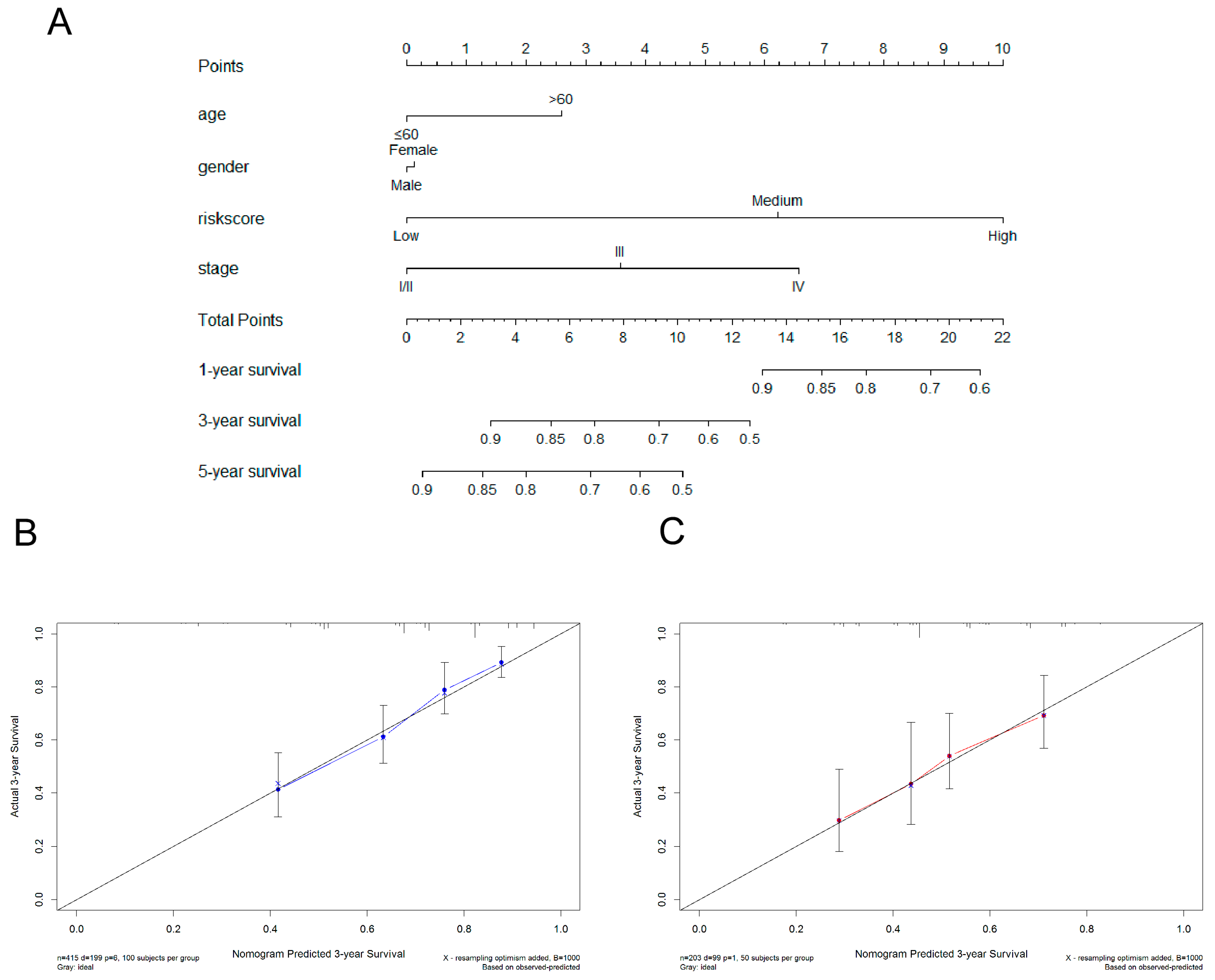
3.6. Construction of Nomogram
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [Green Version]
- Peske, J.D.; Woods, A.B.; Engelhard, V.H. Control of CD8 T-Cell Infiltration into Tumors by Vasculature and Microenvironment. Adv. Cancer Res. 2015, 128, 263–307. [Google Scholar] [PubMed] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Horejsi, V.; Zhang, W.; Schraven, B. Transmembrane adaptor proteins: Organizers of immunoreceptor signalling. Nat. Rev. Immunol. 2004, 4, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Marie-Cardine, A.; Kirchgessner, H.; Bruyns, E.; Shevchenko, A.; Mann, M.; Autschbach, F.; Ratnofsky, S.; Meuer, S.; Schraven, B. SHP2-interacting transmembrane adaptor protein (SIT); a novel disulfide-linked dimer regulating human T cell activation. J. Exp. Med. 1999, 189, 1181–1194. [Google Scholar] [CrossRef] [Green Version]
- Pfrepper, K.I.; Marie-Cardine, A.; Simeoni, L.; Kuramitsu, Y.; Leo, A.; Spicka, J.; Hilgert, I.; Scherer, J.; Schraven, B. Structural and functional dissection of the cytoplasmic domain of the transmembrane adaptor protein SIT (SHP2-interacting transmembrane adaptor protein). Eur. J. Immunol. 2001, 31, 1825–1836. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zhu, Z.; Lan, Y.; Duan, S.; Zhu, Z.; Zhang, X.; Li, G.; Qu, H.; Feng, Y.; Cai, H.; et al. Development and Validation of a CD8+ T Cell Infiltration-Related Signature for Melanoma Patients. Front. Immunol. 2021, 12, 659444. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Kalyana-Sundaram, S.; Mahavisno, V.; Varambally, R.; Yu, J.; Briggs, B.B.; Barrette, T.R.; Anstet, M.J.; Kincead-Beal, C.; Kulkarni, P.; et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007, 9, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemers, N.O.; Holloway, J.L.; Chang, H.; Chasalow, S.D.; Ross-MacDonald, P.B.; Voliva, C.F.; Szustakowski, J.D. Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLoS ONE 2017, 12, e0179726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Maatta, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Y.; Chlewicki, L.K.; Zhang, Y.; Guo, J.; Liang, W.; Wang, J.; Wang, X.; Fu, Y.X. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016, 30, 500. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.; Vignard, V.; Varey, E.; Parrot, T.; Knol, A.C.; Khammari, A.; Gervois, N.; Lang, F.; Dreno, B.; Labarriere, N. Emergence of High-Avidity Melan-A-Specific Clonotypes as a Reflection of Anti-PD-1 Clinical Efficacy. Cancer Res. 2017, 77, 7083–7093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Jia, M. ABCC8 is correlated with immune cell infiltration and overall survival in lower grade glioma. Biocell 2023, 1, 109–123. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. CANCER IMMUNOLOGY. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Albittar, A.A.; Alhalabi, O.; Glitza Oliva, I.C. Immunotherapy for Melanoma. Adv. Exp. Med. Biol. 2020, 1244, 51–68. [Google Scholar]
- Hubener, C.; Mincheva, A.; Lichter, P.; Schraven, B.; Bruyns, E. Complete sequence, genomic organization, and chromosomal localization of the human gene encoding the SHP2-interacting transmembrane adaptor protein (SIT). Immunogenetics 2001, 53, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Arndt, B.; Krieger, T.; Kalinski, T.; Thielitz, A.; Reinhold, D.; Roessner, A.; Schraven, B.; Simeoni, L. The transmembrane adaptor protein SIT inhibits TCR-mediated signaling. PLoS ONE 2011, 6, e23761. [Google Scholar] [CrossRef] [PubMed]
- Buschow, S.I.; van Balkom, B.W.; Aalberts, M.; Heck, A.J.; Wauben, M.; Stoorvogel, W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 2010, 88, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Brill, L.M.; Salomon, A.R.; Ficarro, S.B.; Mukherji, M.; Stettler-Gill, M.; Peters, E.C. Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Anal. Chem. 2004, 76, 2763–2772. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yang, J.; Han, J.; He, J.; Liao, W. A novel immune checkpoint-related seven-gene signature for predicting prognosis and immunotherapy response in melanoma. Int. Immunopharmacol. 2020, 87, 106821. [Google Scholar] [CrossRef]
- Hu, B.; Wei, Q.; Li, X.; Ju, M.; Wang, L.; Zhou, C.; Chen, L.; Li, Z.; Wei, M.; He, M.; et al. Development of an IFNgamma response-related signature for predicting the survival of cutaneous melanoma. Cancer Med. 2020, 9, 8186–8201. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liu, Z.; Liu, X.; Duan, X.; Huang, Y.; Jin, Z.; Niu, Y.; Zhang, L.; Chen, H. Identification of an Immune-Related Prognostic Signature Associated With Immune Infiltration in Melanoma. Front. Genet. 2020, 11, 1002. [Google Scholar] [CrossRef]
- Zhao, Y.; Schaafsma, E.; Gorlov, I.P.; Hernando, E.; Thomas, N.E.; Shen, R.; Turk, M.J.; Berwick, M.; Amos, C.I.; Cheng, C. A Leukocyte Infiltration Score Defined by a Gene Signature Predicts Melanoma Patient Prognosis. Mol. Cancer Res. MCR 2019, 17, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Zeng, Y.; Yin, H.; Chen, F.; Wang, Q.; Yu, X.; Zhou, Y. Exploration of the immune cell infiltration-related gene signature in the prognosis of melanoma. Aging 2021, 13, 3459–3482. [Google Scholar] [CrossRef]
- Zhang, J.A.; Zhou, X.Y.; Huang, D.; Luan, C.; Gu, H.; Ju, M.; Chen, K. Development of an Immune-Related Gene Signature for Prognosis in Melanoma. Front. Oncol. 2020, 10, 602555. [Google Scholar] [CrossRef]
- Egen, J.G.; Ouyang, W.; Wu, L.C. Human Anti-tumor Immunity: Insights from Immunotherapy Clinical Trials. Immunity 2020, 52, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lee, C.K.; Lin, C.H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Hyoung Lee, J.; Hayashi, M.; Dianzani, U.; Ofune, K.; Maruyama, M.; Oe, S.; Ito, T.; Hashiba, T.; Yoshimura, K.; et al. ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro-Oncology 2020, 22, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, Q.; Liao, J.Y.; Song, E.; Xia, Q.; Pan, J.; Li, Y.; Li, J.; Zhou, B.; Ye, Y.; et al. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell 2020, 180, 1081–1097.e24. [Google Scholar] [CrossRef] [PubMed]
- Goudy, K.; Aydin, D.; Barzaghi, F.; Gambineri, E.; Vignoli, M.; Ciullini Mannurita, S.; Doglioni, C.; Ponzoni, M.; Cicalese, M.P.; Assanelli, A.; et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin. Immunol. 2013, 146, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef] [Green Version]
- Tallerico, R.; Garofalo, C.; Carbone, E. A New Biological Feature of Natural Killer Cells: The Recognition of Solid Tumor-Derived Cancer Stem Cells. Front. Immunol. 2016, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Tamada, K.; Shimozaki, K.; Chapoval, A.I.; Zhai, Y.; Su, J.; Chen, S.F.; Hsieh, S.L.; Nagata, S.; Ni, J.; Chen, L. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J. Immunol. 2000, 164, 4105–4110. [Google Scholar] [CrossRef] [Green Version]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Yao, S.; Iliopoulou, B.P.; Han, X.; Augustine, M.M.; Xu, H.; Phennicie, R.T.; Flies, S.J.; Broadwater, M.; Ruff, W.; et al. B7-H5 costimulates human T cells via CD28H. Nat. Commun. 2013, 4, 2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.C.; Han, Q.J.; Ying, H.Y.; Gu, X.X.; Yang, N.; Li, L.Y.; Zhang, Q.Z. TNFSF15 facilitates differentiation and polarization of macrophages toward M1 phenotype to inhibit tumor growth. Oncoimmunology 2022, 11, 2032918. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, M.; Liu, C.; Liu, Y.; Bao, Z.; Jiang, Y.; Sun, X. Discovery and Validation of a SIT1-Related Prognostic Signature Associated with Immune Infiltration in Cutaneous Melanoma. J. Pers. Med. 2023, 13, 13. https://doi.org/10.3390/jpm13010013
Jia M, Liu C, Liu Y, Bao Z, Jiang Y, Sun X. Discovery and Validation of a SIT1-Related Prognostic Signature Associated with Immune Infiltration in Cutaneous Melanoma. Journal of Personalized Medicine. 2023; 13(1):13. https://doi.org/10.3390/jpm13010013
Chicago/Turabian StyleJia, Ming, Chengfei Liu, Yuean Liu, Zhengqiang Bao, Yuhua Jiang, and Xifeng Sun. 2023. "Discovery and Validation of a SIT1-Related Prognostic Signature Associated with Immune Infiltration in Cutaneous Melanoma" Journal of Personalized Medicine 13, no. 1: 13. https://doi.org/10.3390/jpm13010013
APA StyleJia, M., Liu, C., Liu, Y., Bao, Z., Jiang, Y., & Sun, X. (2023). Discovery and Validation of a SIT1-Related Prognostic Signature Associated with Immune Infiltration in Cutaneous Melanoma. Journal of Personalized Medicine, 13(1), 13. https://doi.org/10.3390/jpm13010013