
Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence and Structure Retrieval
2.2. Prediction of Linear B-Cell Epitopes
2.3. Prediction of Helper T-Lymphocyte (HTL) Epitopes
2.4. Prediction of Cytotoxic T-Lymphocyte (CTL) Epitopes
2.5. Selection of Adjuvants and Guaranty Linkers for Vaccine Construction
2.6. Toxicity Detection, Solubility Prediction, and Physicochemical Properties of Predicted Vaccine Structures
2.7. Prediction of Immunogenicity, Antigenicity, and Sensitization of Vaccines
2.8. Secondary Structure and 3D Structure Prediction, Optimization, and Verification
2.9. Prediction of Discontinuous B-Cell Epitopes
2.10. Candidate Vaccine Docked to Toll-like Receptor (TLR) 4 Molecules
2.11. Immune Simulation
2.12. Normal Mode Analysis (NMA) of the Complex
2.13. Codon Optimization
3. Results
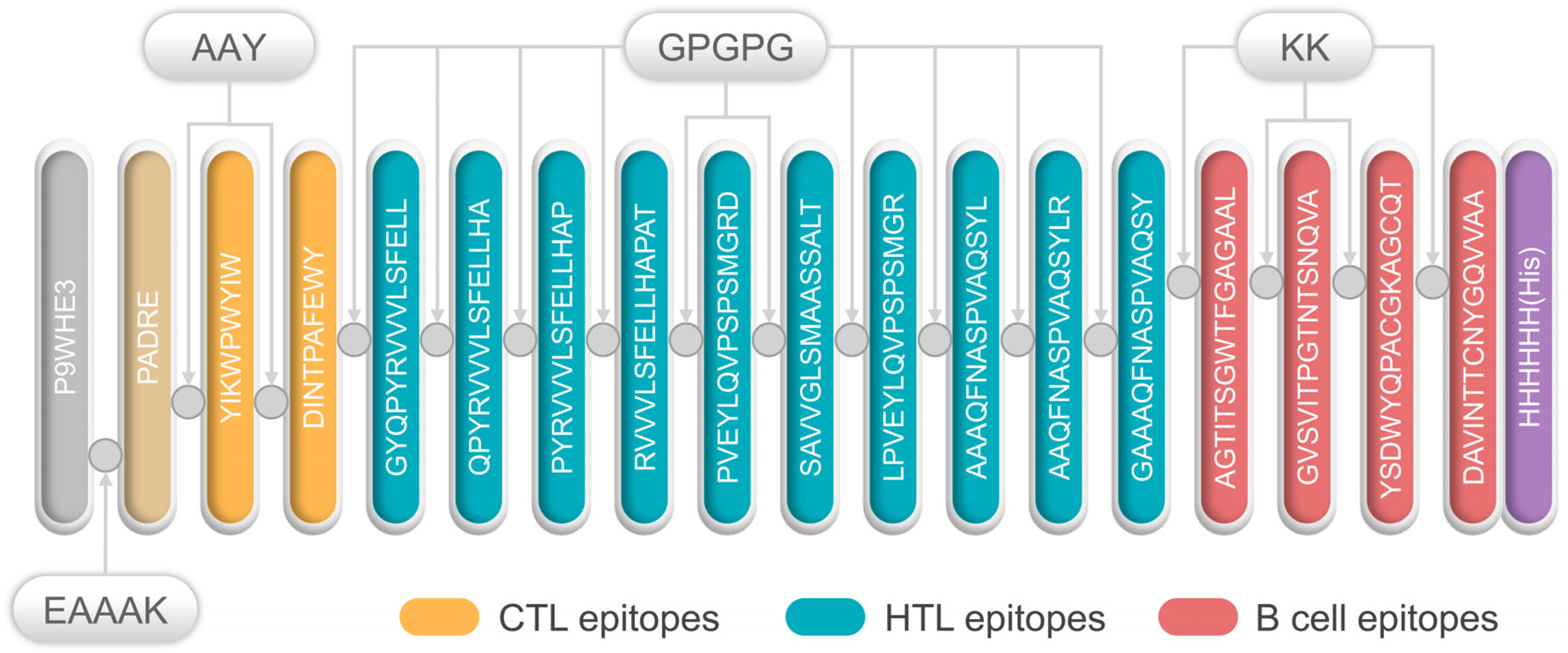
3.1. Selection of Immunodominant Epitopes
3.2. Primary Structure Construction of the S7D5L4 Vaccine
3.3. Toxicity Detection, Solubility Prediction, and Physicochemical Properties of the S7D5L4 Vaccine
3.4. Immunogenicity, Antigenicity, and Sensitization of the S7D5L4 Vaccine
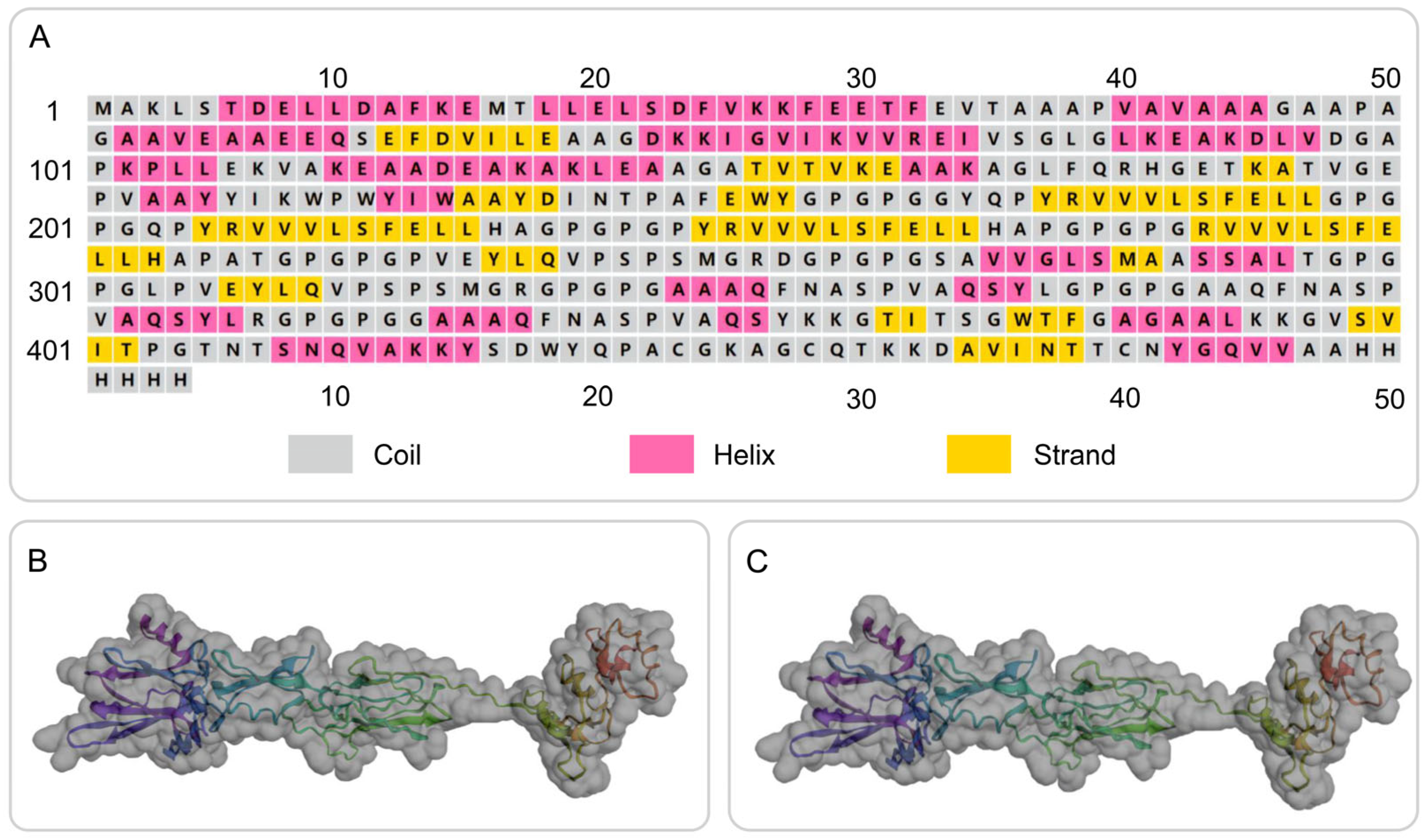
3.5. Secondary Structure Prediction and 3D Structure Modeling of the S7D5L4 Vaccine
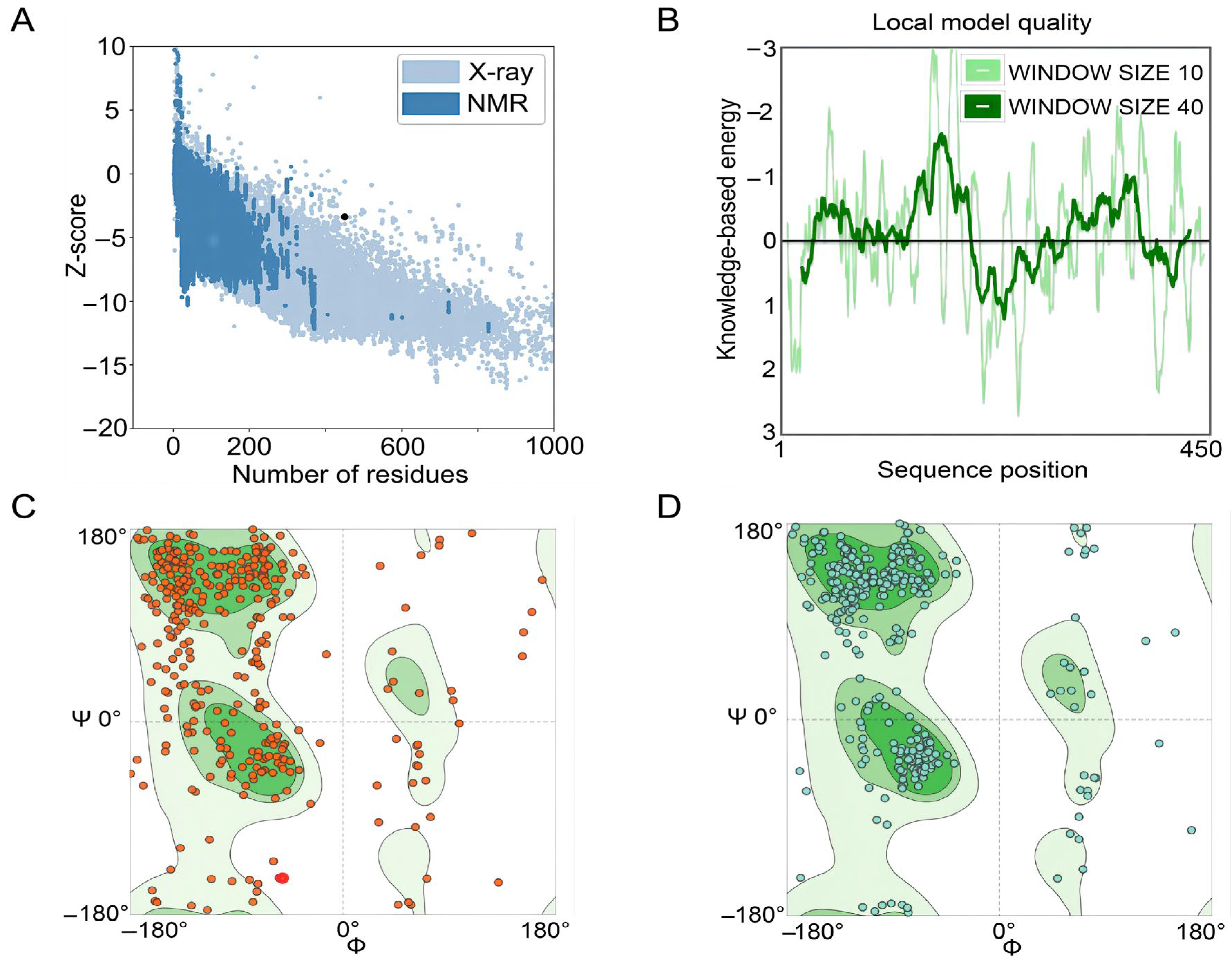
3.6. Optimization of the Tertiary Structure of the S7D5L4 Vaccine
3.7. Discontinuous B-Cell Epitopes
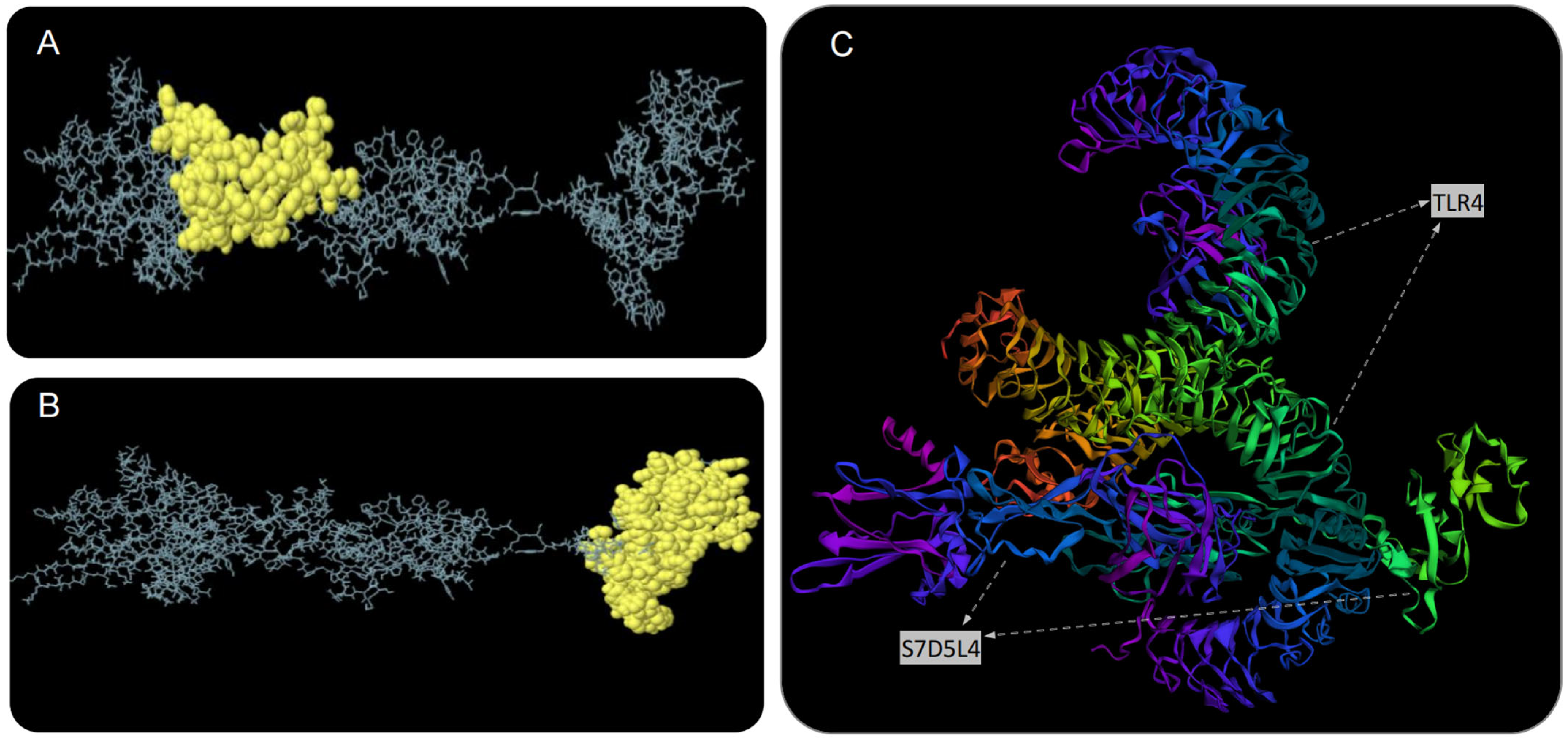
3.8. The S7D5L4 Vaccine Docked to TLR4 Molecule
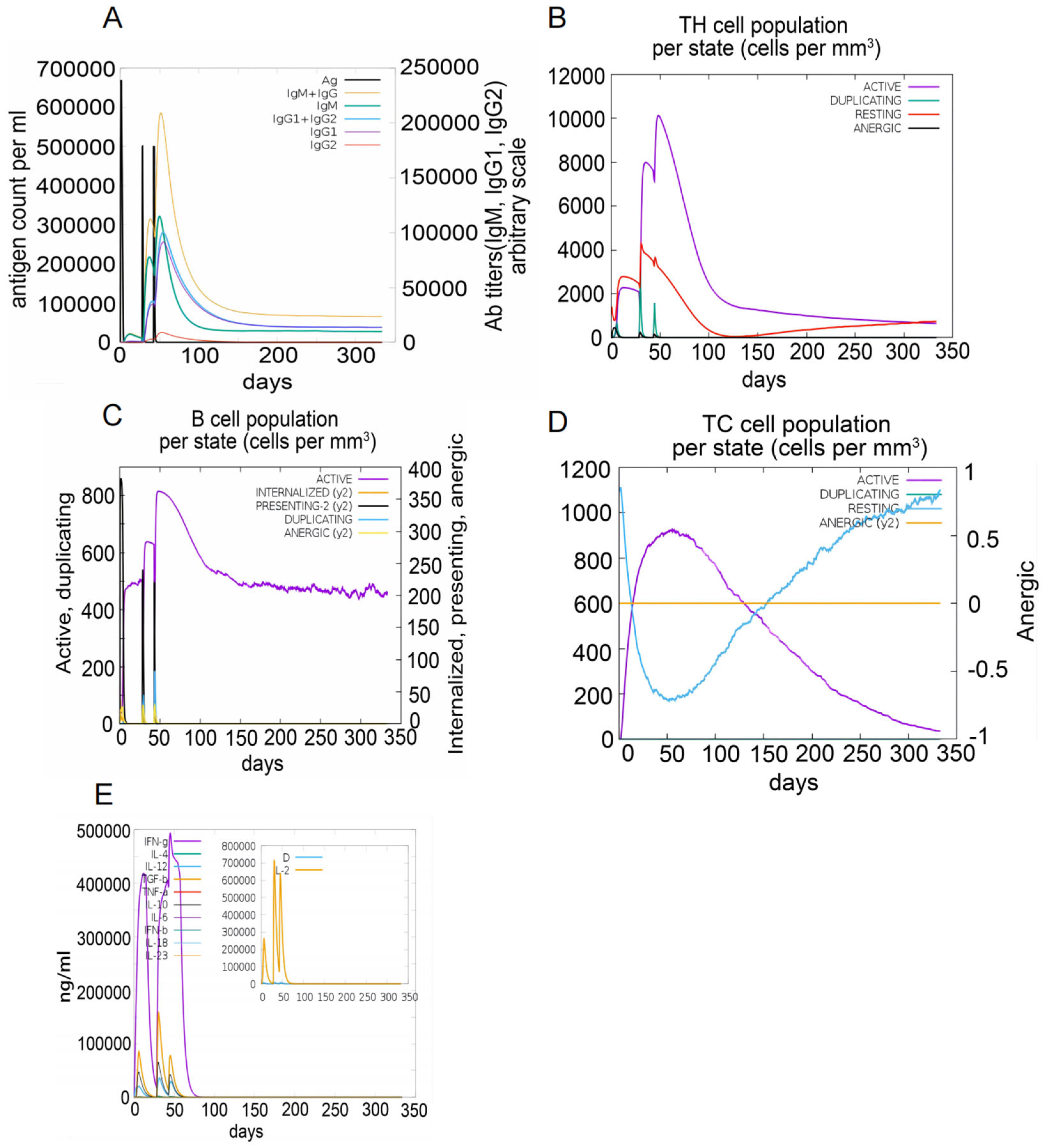
3.9. Immune Simulation of the S7D5L4 Vaccine
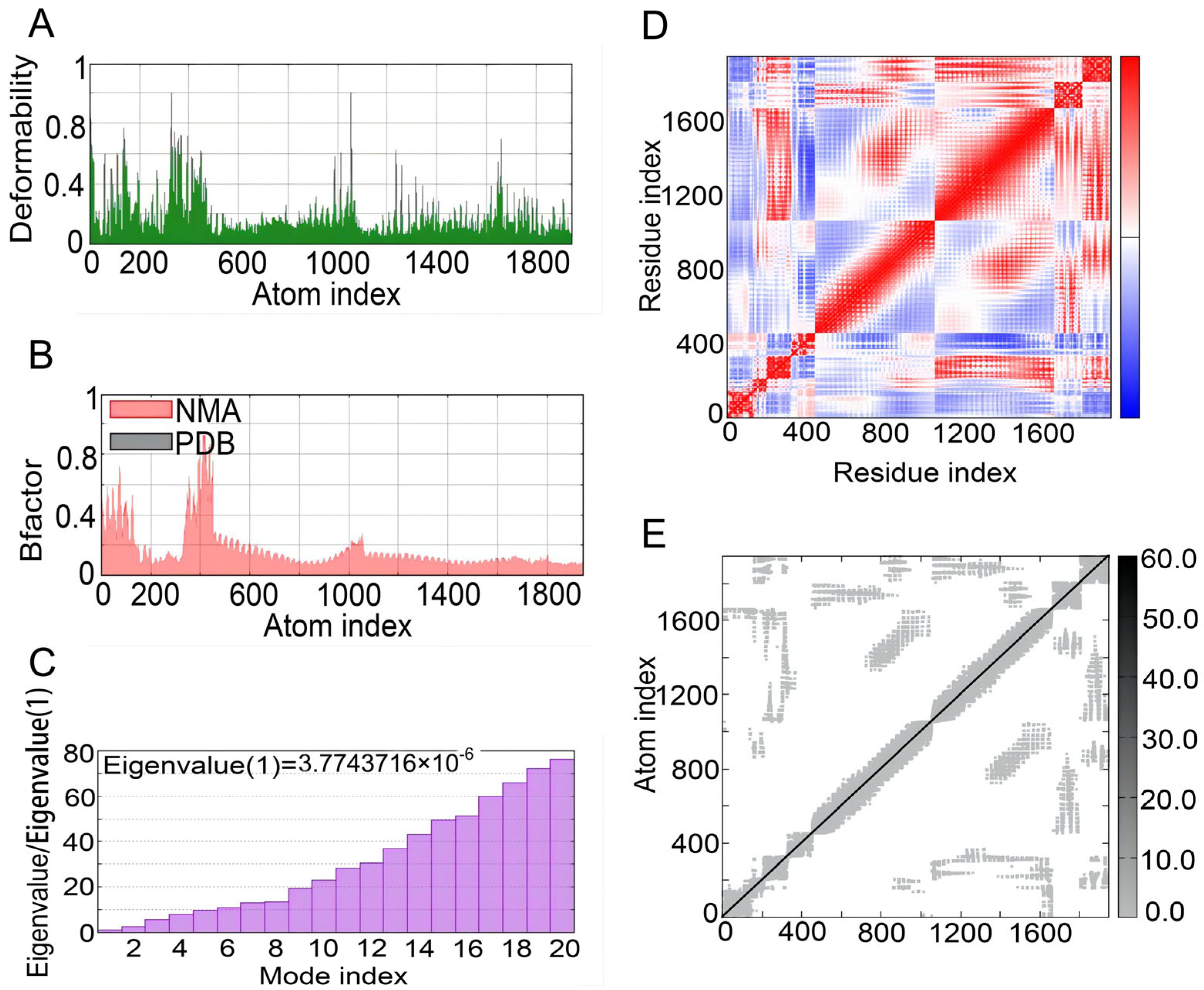
3.10. NMA of the Complex
3.11. Codon Optimization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gong, W.; Pan, C.; Cheng, P.; Wang, J.; Zhao, G.; Wu, X. Peptide-Based Vaccines for Tuberculosis. Front. Immunol. 2022, 13, 830497. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liang, Y.; Wu, X. The current status, challenges, and future developments of new tuberculosis vaccines. Hum. Vaccines Immunother. 2018, 14, 1697–1716. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022.
- Bostanghadiri, N.; Jazi, F.M.; Razavi, S.; Fattorini, L.; Darban-Sarokhalil, D. Mycobacterium tuberculosis and SARS-CoV-2 Coinfections: A Review. Front. Microbiol. 2021, 12, 747827. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Shah, Z.; Yasmeen, N.; Baloch, Z.; Xia, X. Pathogenesis of SARS-CoV-2 and Mycobacterium tuberculosis Coinfection. Front. Immunol. 2022, 13, 909011. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Centis, R.; D’Ambrosio, L.; García-García, J.M.; Goletti, D.; Gualano, G.; Lipani, F.; Palmieri, F.; Sánchez-Montalvá, A.; Pontali, E.; et al. Tuberculosis, COVID-19 and migrants: Preliminary analysis of deaths occurring in 69 patients from two cohorts. Pulmonology 2020, 26, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Adzic-Vukicevic, T.; Stosic, M.; Antonijevic, G.; Jevtic, M.; Radovanovic-Spurnic, A.; Velickovic, J. Tuberculosis and COVID-19 co-infection in Serbia: Pandemic challenge in a low-burden country. Front. Med. 2022, 9, 971008. [Google Scholar] [CrossRef]
- Li, W.; Li, M.; Deng, G.; Zhao, L.; Liu, X.; Wang, Y. Prime-boost vaccination with Bacillus Calmette Guerin and a recombinant adenovirus co-expressing CFP10, ESAT6, Ag85A and Ag85B of Mycobacterium tuberculosis induces robust antigen-specific immune responses in mice. Mol. Med. Rep. 2015, 12, 3073–3080. [Google Scholar] [CrossRef]
- Tkachuk, A.P.; Bykonia, E.N.; Popova, L.I.; Kleymenov, D.A.; Semashko, M.A.; Chulanov, V.P.; Fitilev, S.B.; Maksimov, S.L.; Smolyarchuk, E.A.; Manuylov, V.A.; et al. Safety and Immunogenicity of the GamTBvac, the Recombinant Subunit Tuberculosis Vaccine Candidate: A Phase II, Multi-Center, Double-Blind, Randomized, Placebo-Controlled Study. Vaccines 2020, 8, 652. [Google Scholar] [CrossRef]
- Liu, X.; Peng, J.; Hu, L.; Luo, Y.; Niu, H.; Bai, C.; Wang, Q.; Li, F.; Yu, H.; Wang, B.; et al. A multistage Mycobacterium tuberculosis subunit vaccine LT70 including latency antigen Rv2626c induces long-term protection against tuberculosis. Hum. Vaccines Immunother. 2016, 12, 1670–1677. [Google Scholar] [CrossRef]
- Evans, J.T.; Ward, J.R.; Kern, J.; Johnson, M.E. A single vaccination with protein-microspheres elicits a strong CD8 T-cell-mediated immune response against Mycobacterium tuberculosis antigen Mtb8.4. Vaccine 2004, 22, 1964–1972. [Google Scholar] [CrossRef]
- Li, H.; Li, R.; Zhong, S.; Ren, H. Plasmid encoding human IL-12 improve protective efficacy of Mtb8.4 gene vaccine with signal sequence against infection of Mycobacterium tuberculosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2007, 23, 291–294. [Google Scholar]
- Luo, Y.; Wang, B.; Hu, L.; Yu, H.; Da, Z.; Jiang, W.; Song, N.; Qie, Y.; Wang, H.; Tang, Z.; et al. Fusion protein Ag85B-MPT64(190-198)-Mtb8.4 has higher immunogenicity than Ag85B with capacity to boost BCG-primed immunity against Mycobacterium tuberculosis in mice. Vaccine 2009, 27, 6179–6185. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Jia, Z.; Gong, W. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 2021, 36, e124. [Google Scholar] [CrossRef]
- Gong, W.; Aspatwar, A.; Wang, S.; Parkkila, S.; Wu, X. COVID-19 pandemic: SARS-CoV-2 specific vaccines and challenges, protection via BCG trained immunity, and clinical trials. Expert Rev. Vaccines 2021, 20, 857–880. [Google Scholar] [CrossRef]
- Nabel, G.J. HIV vaccine strategies. Vaccine 2002, 20, 1945–1947. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Immunoinformatics: A brief review. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2014; Volume 1184, pp. 23–55. [Google Scholar] [CrossRef]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and Vaccine Development: An Overview. Immunotargets Ther. 2020, 9, 13–30. [Google Scholar] [CrossRef]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020, 92, 495–500. [Google Scholar] [CrossRef]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2021, 39, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, L.; Gong, W. In silico Analysis of Peptide-Based Biomarkers for the Diagnosis and Prevention of Latent Tuberculosis Infection. Front. Microbiol. 2022, 13, 947852. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Gong, W.; Liang, Y.; Wu, X.; Zhao, W. Prediction and analyses of HLA-II restricted Mycobacterium tuberculosis CD4(+) T cell epitopes in the Chinese population. Biotechnol. Appl. Biochem. 2022, 69, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Mehta, S.; Bharrhan, S.; Chen, Y.; Achkar, J.M.; Casadevall, A.; Flynn, J. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin. Immunol. 2014, 26, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E.; et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar] [CrossRef]
- Ayyagari, V.S.; Venkateswarulu, T.C.; Abraham, P.K.; Srirama, K. Design of a multi-epitope-based vaccine targeting M-protein of SARS-CoV2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2022, 40, 2963–2977. [Google Scholar] [CrossRef]
- Bastola, R.; Noh, G.; Keum, T.; Bashyal, S.; Seo, J.E.; Choi, J.; Oh, Y.; Cho, Y.; Lee, S. Vaccine adjuvants: Smart components to boost the immune system. Arch. Pharm. Res. 2017, 40, 1238–1248. [Google Scholar] [CrossRef]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Garnier, J.; Gibrat, J.F.; Robson, B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996, 266, 540–553. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. An Overview of Recent Insights into the Response of TLR to SARS-CoV-2 Infection and the Potential of TLR Agonists as SARS-CoV-2 Vaccine Adjuvants. Viruses 2021, 13, 2302. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Al-Hawash, A.B.; Zhang, X.; Ma, F. Strategies of codon optimization for high-level heterologous protein expression in microbial expression systems. Gene Rep. 2017, 9, 46–53. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef]
- Jayaweera, M.; Perera, H.; Gunawardana, B.; Manatunge, J. Transmission of COVID-19 virus by droplets and aerosols: A critical review on the unresolved dichotomy. Environ. Res. 2020, 188, 109819. [Google Scholar] [CrossRef]
- Mirzaei, R.; Goodarzi, P.; Asadi, M.; Soltani, A.; Aljanabi, H.A.A.; Jeda, A.S.; Dashtbin, S.; Jalalifar, S.; Mohammadzadeh, R.; Teimoori, A.; et al. Bacterial co-infections with SARS-CoV-2. IUBMB Life 2020, 72, 2097–2111. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A.; Namvar, A. An overview of in silico vaccine design against different pathogens and cancer. Expert Rev. Vaccines 2020, 19, 699–726. [Google Scholar] [CrossRef]
- Kapingidza, A.B.; Kowal, K.; Chruszcz, M. Antigen-Antibody Complexes. Subcell. Biochem. 2020, 94, 465–497. [Google Scholar] [CrossRef]
- Lee, S.; Nguyen, M.T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015, 15, 51–57. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. Publ. Protein Soc. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Pati, P.K.; Pati, A.M.; Nagpal, A.K. Physico-chemical characterization and topological analysis of pathogenesis-related proteins from Arabidopsis thaliana and Oryza sativa using in-silico approaches. PLoS ONE 2020, 15, e0239836. [Google Scholar] [CrossRef] [PubMed]
- Nehete, J.Y.; Bhambar, R.S.; Narkhede, M.R.; Gawali, S.R. Natural proteins: Sources, isolation, characterization and applications. Pharmacogn. Rev. 2013, 7, 107–116. [Google Scholar] [CrossRef]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 49, 309–317. [Google Scholar] [CrossRef]
- Lim, H.X.; Lim, J.; Jazayeri, S.D.; Poppema, S.; Poh, C.L. Development of multi-epitope peptide-based vaccines against SARS-CoV-2. Biomed. J. 2021, 44, 18–30. [Google Scholar] [CrossRef]
- de Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef]
- Panagioti, E.; Klenerman, P.; Lee, L.N.; van der Burg, S.H.; Arens, R. Features of Effective T Cell-Inducing Vaccines against Chronic Viral Infections. Front. Immunol. 2018, 9, 276. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Peptide Sequence | Length | Alleles | Percentile Rank a | Antigenicity Score b | IFN-γ Score c | Immunogenicity Score d | ABC Pred Score e | AllerTOP V 1.0 | AllergenFP v.2.0 | Toxin Pred |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HTL epitopes | |||||||||||
| S protein | |||||||||||
| GYQPYRVVVLSFELL | 15 | HLA-DPA1*01:03/DPB1*02:01 | 0.36 | 1.074 | 0.6533982 | Non f | Non f | Non f | |||
| QPYRVVVLSFELLHA | 15 | HLA-DPA1*01:03/DPB1*02:01 | 0.36 | 0.9109 | 0.60855322 | Non | Non | Non | |||
| PYRVVVLSFELLHAP | 15 | HLA-DPA1*03:01/DPB1*04:02 | 0.25 | 0.8161 | 0.56872818 | Non | Non | Non | |||
| RVVVLSFELLHAPAT | 15 | HLA-DRB1*01:01 | 0.24 | 0.7485 | 0.5092653 | Non | Non | Non | |||
| Ag85a | |||||||||||
| PVEYLQVPSPSMGRD | 15 | HLA-DRB1*04:01 | 0.46 | 0.7094 | 0.99952242 | Non | Non | Non | |||
| SAVVGLSMAASSALT | 15 | HLA-DRB1*09:01 | 0.34 | 0.6602 | 0.86704792 | Non | Non | Non | |||
| LPVEYLQVPSPSMGR | 15 | HLA-DRB1*04:01 | 0.38 | 0.7922 | 0.82782141 | Non | Non | Non | |||
| Mtb8.4 | |||||||||||
| AAAQFNASPVAQSYL | 15 | HLA-DRB1*09:01 | 0.34 | 0.6839 | 0.45466067 | Non | Non | Non | |||
| AAQFNASPVAQSYLR | 15 | HLA-DRB1*09:01 | 0.34 | 0.6176 | 0.56966616 | Non | Non | Non | |||
| GAAAQFNASPVAQSY | 15 | HLA-DRB1*09:01 | 0.35 | 0.5933 | 0.39892204 | Non | Non | Non | |||
| CTL epitopes | |||||||||||
| S protein | |||||||||||
| YIKWPWYIW | 9 | HLA-A*23:01 | 0.1 | 0.9673 | 0.42524 | Non | Non | Non | |||
| Ag85a | |||||||||||
| DINTPAFEWY | 10 | HLA-A*26:01 | 0.24 | 1.8593 | 0.38838 | Non | Non | Non | |||
| B-cell epitopes | |||||||||||
| S protein | |||||||||||
| AGTITSGWTFGAGAAL | 16 | 0.97 | Non | Non | Non | ||||||
| GVSVITPGTNTSNQVA | 16 | 0.95 | Non | Non | Non | ||||||
| Ag85a | |||||||||||
| YSDWYQPACGKAGCQT | 16 | 0.93 | Non | Non | Non | ||||||
| Mtb8.4 | |||||||||||
| DAVINTTCNYGQVVAA | 16 | 0.86 | Non | Non | Non | ||||||
| Parameters | Results | |
|---|---|---|
| Biological characteristics | ||
| Antigenicity | 0.7811 a | |
| 0.4299 b | ||
| Immunogenicity | 1.45499 | |
| Half-life (h) c | ||
| Mammalian reticulocytes (in vitro) | >30 | |
| Yeast (in vivo) | >20 | |
| E. coli (in vivo) | >10 | |
| Physicochemical properties | ||
| Molecular weight (Da) | 46,825.3 | |
| Number of amino acids | 454 | |
| Isoelectric point | 6.38 | |
| Instability index | 26.51 | |
| Fat index | 80.24 | |
| Basic features | ||
| Toxicity | Non-toxic | |
| Sensitization | Non-allergenic | |
| Solubility | 0.462 | |
| GRAVY score | −0.033 | |
| No | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A:G331, A:P332, A:G333, A:A341, A:A345, A:Q346, A:S347, A:A349, A:A350, A:Y351, A:Y352, A:I353, A:K354, A:W355, A:P356, A:W357, A:Y358, A:I359, A:W360, A:A361, A:A362, A:Y363, A:I365, A:N366, A:T367, A:P368, A:A369, A:F370, A:E371, A:Y373, A:K374, A:K375, A:G376, A:T377, A:I378, A:T379, A:S380, A:G381, A:W382, A:T383, A:F384, A:G385, A:A386, A:G387, A:A388, A:A389, A:L390, A:K391, A:G393, A:V394, A:S395, A:V396, A:I397, A:T398, A:P399, A:G400, A:T401, A:N402, A:T403, A:S404, A:N405, A:Q406, A:V407, A:A408, A:K409, A:K410, A:Y411, A:S412, A:D413, A:Y415, A:Q416, A:P417, A:A418, A:C419, A:G420, A:K421, A:A422, A:G423, A:C424, A:Q425, A:T426, A:K427, A:K428, A:D429, A:A430, A:V431, A:I432, A:N433, A:T434, A:T435, A:C436, A:N437, A:Y438, A:G439, A:Q440, A:V441, A:V442, A:A443, A:A444, A:H445, A:H446, A:H447, A:H448, A:H449, A:H450 | 105 | 0.792 # |
| 2 | A:Q135, A:H136, A:H137, A:G138, A:E139, A:G140, A:A143, A:T144, A:V145, A:G146, A:E147, A:P148, A:V149, A:E150, A:K151, A:Y158, A:V160, A:V161, A:V162, A:L163, A:G169, A:P170, A:G171, A:P172, A:G173, A:Q174, A:P175, A:Y176, A:R177, A:V178, A:V179, A:V180, A:L181, A:S182, A:F183, A:E184, A:L185, A:L186, A:H187, A:A188, A:G189, A:P190, A:G191, A:P192, A:G193, A:P194, A:Y195, A:R196, A:V197, A:V198, A:V199, A:L200, A:S201, A:F202, A:E203, A:L204, A:L205 | 84 | 0.693 # |
| Cluster | Members | Representative | Weighted Score |
|---|---|---|---|
| 0 | 27 | Center | −921.5b |
| Lowest Energy | −1208.1b | ||
| 1 | 27 | Center | −1004 |
| Lowest Energy | −1072.3 | ||
| 3 | 20 | Center | −962.3 |
| Lowest Energy | −1058.4 | ||
| 4 | 16 | Center | −1057.3 |
| Lowest Energy | −1057.3 | ||
| 11 | 13 | Center | −883.5 |
| Lowest Energy | −1031.6 | ||
| 13 | 12 | Center | −932.2 |
| Lowest Energy | −1114.5 | ||
| 14 | 12 | Center | −1088.4 |
| Lowest Energy | −1143.5 | ||
| 15 | 12 | Center | −995.9 |
| Lowest Energy | −1040.1 | ||
| 22 | 10 | Center | −1014.6 |
| Lowest Energy | −1014.6 | ||
| 29 | 9 | Center | −1089 |
| Lowest Energy | −1089 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, C.; Tang, F.; Wang, J.; Cheng, P.; Wang, L.; Gong, W. Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2. J. Pers. Med. 2023, 13, 116. https://doi.org/10.3390/jpm13010116
Peng C, Tang F, Wang J, Cheng P, Wang L, Gong W. Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2. Journal of Personalized Medicine. 2023; 13(1):116. https://doi.org/10.3390/jpm13010116
Chicago/Turabian StylePeng, Cong, Fengjie Tang, Jie Wang, Peng Cheng, Liang Wang, and Wenping Gong. 2023. "Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2" Journal of Personalized Medicine 13, no. 1: 116. https://doi.org/10.3390/jpm13010116
APA StylePeng, C., Tang, F., Wang, J., Cheng, P., Wang, L., & Gong, W. (2023). Immunoinformatic-Based Multi-Epitope Vaccine Design for Co-Infection of Mycobacterium tuberculosis and SARS-CoV-2. Journal of Personalized Medicine, 13(1), 116. https://doi.org/10.3390/jpm13010116