
Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities


Abstract

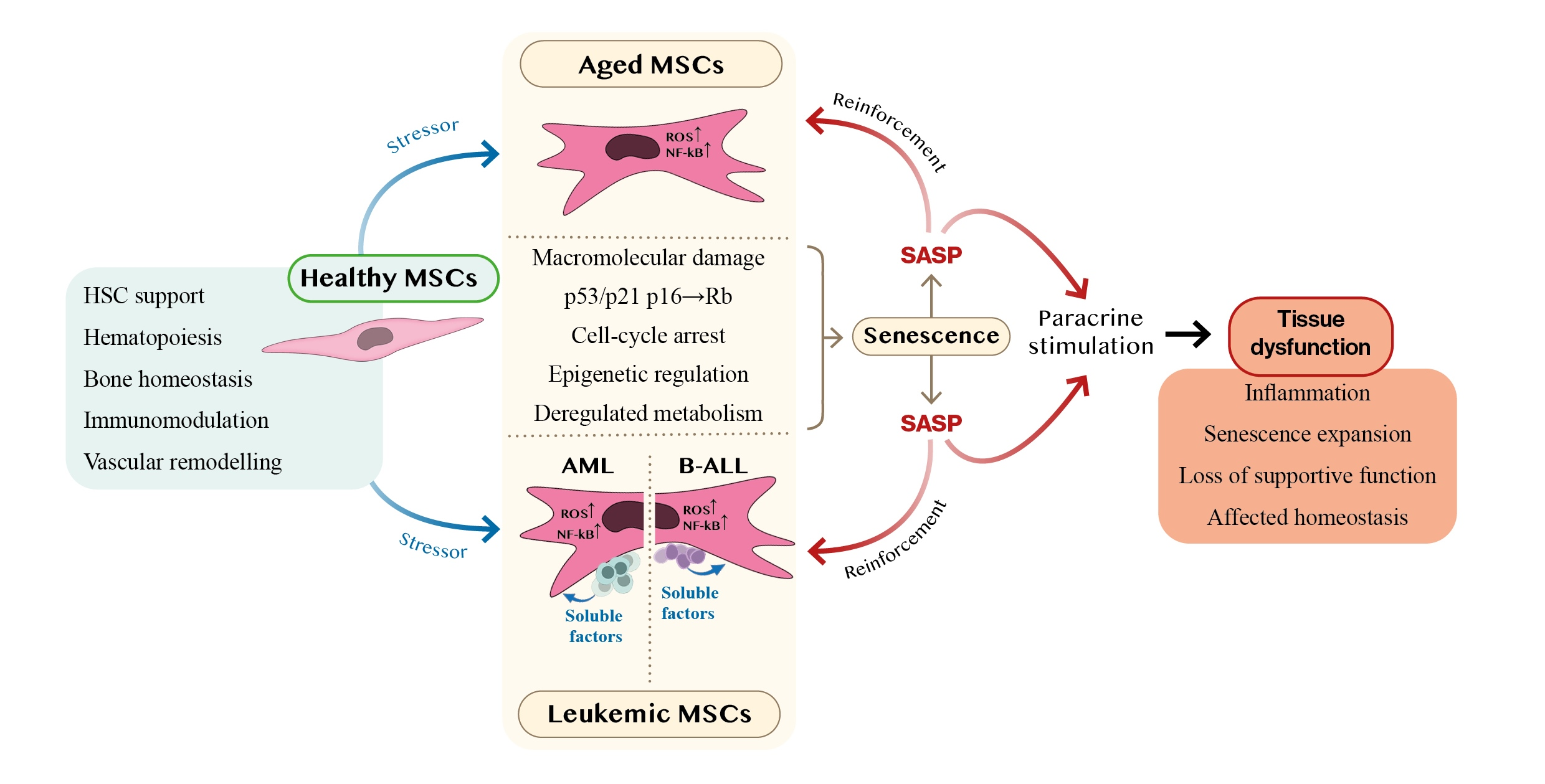{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bone Marrow Aging and Cellular Dysfunctions
3. The Aging and Senescence of Mesenchymal Stem Cells
4. The Effect of Leukemic Cells on Mesenchymal Stem Cell Properties
4.1. Cellular Dysfunctions of MSCs in Aging and Senescence
4.1.1. Cellular Dysfunction of MSCs in AML
4.1.2. The Cellular Dysfunction of MSCs in B-ALL
4.2. The Redox Balance of MSCs in Aging and Senescence
4.2.1. Redox Balance of MSCs in AML
4.2.2. Redox Balance of MSCs in B-ALL
4.3. Genetic, Epigenetic and Gene Expression Alterations of MSCs in Aging and Senescence
4.3.1. Genetic, Epigenetic and Gene Expression Alterations of MSCs in AML
4.3.2. Genetic, Epigenetic and Gene Expression Alterations of MSCs in B-ALL
4.4. SASP and Pro-Inflammatory Stimulation in Aging and Senescence
4.4.1. SASP and Pro-Inflammatory Stimulation in AML
4.4.2. SASP and Pro-Inflammatory Stimulation in B-ALL
4.5. Stemness Property Modifications of MSCs in Aging and Senescence
4.5.1. Stemness Properties Modifications of MSCs in AML
4.5.2. Stemness Property Modifications of MSCs in B-ALL
5. MSC Roles in Drug Resistance
5.1. Drug Resistance Mechanisms in AML
5.2. Drug Resistance in B-ALL
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, S.; D’Adda di Fagagna, F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell 2012, 11, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Idda, M.L.; Mcclusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and tissue-specific expression of senescence biomarkers in mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef]
- Verovskaya, E.V.; Dellorusso, P.V.; Passegué, E. Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med. 2019, 25, 494–515. [Google Scholar] [CrossRef]
- Woods, K.; Guezguez, B. Dynamic Changes of the Bone Marrow Niche: Mesenchymal Stromal Cells and Their Progeny During Aging and Leukemia. Front. Cell Dev. Biol. 2021, 9, 2228. [Google Scholar] [CrossRef]
- Civini, S.; Jin, P.; Ren, J.; Sabatino, M.; Castiello, L.; Jin, J.; Wang, H.; Zhao, Y.; Marincola, F.; Stroncek, D. Leukemia cells induce changes in human bone marrow stromal cells. J. Transl. Med. 2013, 11, 298. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.; Rodríguez, C.I. Cell and Cell-Free Therapies to Counteract Human Premature and Physiological Aging: MSCs Come to Light. J. Pers. Med. 2021, 11, 1043. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.G.P. Niche contributions to oncogenesis: Emerging concepts and implications for the hematopoietic system. Haematologica 2011, 96, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xia, X.; Li, B. Mesenchymal stem cell aging: Mechanisms and influences on skeletal and non-skeletal tissues. Exp. Biol. Med. 2015, 240, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Saraber, P.; Ding, Z.; Kusumbe, A.P. Diversity of Vascular Niches in Bones and Joints During Homeostasis, Ageing, and Diseases. Front. Immunol. 2021, 12, 5252. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; Haan, G. De Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Gnani, D.; Crippa, S.; della Volpe, L.; Rossella, V.; Conti, A.; Lettera, E.; Rivis, S.; Ometti, M.; Fraschini, G.; Bernardo, M.E.; et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 2019, 18, e12933. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, G.; Schiller, P.C.; Ricordi, C.; Roos, B.A.; Howard, G.A. Age-Related Osteogenic Potential of Mesenchymal Stromal Stem Cells from Human Vertebral Bone Marrow. J. Bone Miner. Res. 1999, 14, 1115–1122. [Google Scholar] [CrossRef]
- Kretlow, J.D.; Jin, Y.Q.; Liu, W.; Zhang, W.J.; Hong, T.H.; Zhou, G.; Baggett, L.S.; Mikos, A.G.; Cao, Y. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008, 9, 60. [Google Scholar] [CrossRef]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Ageing Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef]
- Demontiero, O.; Vidal, C.; Duque, G. Aging and bone loss: New insights for the clinician. Ther. Adv. Musculoskelet. Dis. 2012, 4, 61–76. [Google Scholar] [CrossRef]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef]
- Muruganandan, S.; Ionescu, A.M.; Sinal, C.J. At the Crossroads of the Adipocyte and Osteoclast Differentiation Programs: Future Therapeutic Perspectives. Int. J. Mol. Sci. 2020, 21, 2277. [Google Scholar] [CrossRef]
- Bianco, P. “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 677–704. [Google Scholar] [CrossRef]
- Belyavsky, A.; Petinati, N.; Drize, N. Hematopoiesis during Ontogenesis, Adult Life, and Aging. Int. J. Mol. Sci. 2021, 22, 9231. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mäe, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Poulos, M.G.; Ramalingam, P.; Gutkin, M.C.; Llanos, P.; Gilleran, K.; Rabbany, S.Y.; Butler, J.M. Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J. Clin. Investig. 2017, 127, 4163–4178. [Google Scholar] [CrossRef]
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol. 2017, 106, 45–54. [Google Scholar] [CrossRef]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef]
- McElhaney, J.E.; Effros, R.B. Immunosenescence: What does it mean to health outcomes in older adults? Curr. Opin. Immunol. 2009, 21, 418–424. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Udupa, K.B.; Xiao, H.; Lipschitz, D.A. Effect of age on marrow macrophage number and function. Aging 1995, 7, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Signer, R.A.J.; Montecino-Rodriguez, E.; Witte, O.N.; McLaughlin, J.; Dorshkind, K. Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 2007, 110, 1831–1839. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.T.; Purton, L.E. A Microenvironment-Induced Myeloproliferative Syndrome Caused by Retinoic Acid Receptor γ Deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef]
- Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Song, R.; Shin, J.; Jeong, D.C.; Kim, S.H.; Kong, Y.Y. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008, 112, 4628–4638. [Google Scholar] [CrossRef]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef]
- Zimmer, S.N.; Lemieux, M.E.; Karia, B.P.; Day, C.; Zhou, T.; Zhou, Q.; Kung, A.L.; Suresh, U.; Chen, Y.; Kinney, M.C.; et al. Mice heterozygous for CREB binding protein are hypersensitive to γ-radiation and invariably develop myelodysplastic/myeloproliferative neoplasm. Exp. Hematol. 2012, 40, 295–306.e5. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar]
- Dong, L.; Yu, W.M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef]
- Vas, V.; Senger, K.; Dörr, K.; Niebel, A.; Geiger, H. Aging of the Microenvironment Influences Clonality in Hematopoiesis. PLoS ONE 2012, 7, e42080. [Google Scholar] [CrossRef]
- Yao, J.C.; Link, D.C. Concise Review: The Malignant Hematopoietic Stem Cell Niche. Stem Cells 2017, 35, 3–8. [Google Scholar] [CrossRef]
- Dander, E.; Palmi, C.; D’amico, G.; Cazzaniga, G. The bone marrow niche in b-cell acute lymphoblastic leukemia: The role of microenvironment from pre-leukemia to overt leukemia. Int. J. Mol. Sci. 2021, 22, 4426. [Google Scholar]
- Bigildeev, A.E.; Pilunov, A.M.; Sats, N.V.; Surin, V.L.; Shipounova, I.N.; Petinati, N.A.; Logacheva, M.D.; Fedotova, A.V.; Kasyanov, A.S.; Artyukhov, A.S.; et al. Clonal Composition of Human Multipotent Mesenchymal Stromal Cells: Application of Genetic Barcodes in Research. Biochemistry 2019, 84, 250–262. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal stem cell: Keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012, 6, 2853–2868. [Google Scholar] [CrossRef]
- Vernot, J.P. Senescence-Associated Pro-inflammatory Cytokines and Tumor Cell Plasticity. Front. Mol. Biosci. 2020, 7, 63. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Zhou, S.; Greenberger, J.S.; Epperly, M.W.; Goff, J.P.; Adler, C.; Leboff, M.S.; Glowacki, J. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 2008, 7, 335–343. [Google Scholar] [CrossRef]
- Baker, N.; Boyette, L.B.; Tuan, R.S. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone 2015, 70, 37–47. [Google Scholar] [CrossRef]
- Caplan, A.I. Review: Mesenchymal stem cells: Cell-based reconstructive therapy in orthopedics. Tissue Eng. 2005, 11, 1198–1211. [Google Scholar] [CrossRef]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and Replicative Senescence Have Related Effects on Human Stem and Progenitor Cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef]
- Han, J.; Mistriotis, P.; Lei, P.; Wang, D.; Liu, S.; Andreadis, S.T. Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells 2012, 30, 2746–2759. [Google Scholar] [CrossRef]
- Boyette, L.; Tuan, R. Adult Stem Cells and Diseases of Aging. J. Clin. Med. 2014, 3, 88–134. [Google Scholar] [CrossRef]
- Neri, S.; Borzì, R.M. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules 2020, 10, 340. [Google Scholar] [CrossRef]
- Jeremy Wen, Q.; Yang, Q.; Goldenson, B.; Malinge, S.; Lasho, T.; Schneider, R.K.; Breyfogle, L.J.; Schultz, R.; Gilles, L.; Koppikar, P.; et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat. Med. 2015, 21, 1473–1480. [Google Scholar] [CrossRef]
- Abe-Suzuki, S.; Kurata, M.; Abe, S.; Onishi, I.; Kirimura, S.; Nashimoto, M.; Murayama, T.; Hidaka, M.; Kitagawa, M. CXCL12+ stromal cells as bone marrow niche for CD34+ hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Lab. Investig. 2014, 94, 1212–1223. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2015, 30, 683–691. [Google Scholar] [CrossRef]
- Vicente López, Á.; Vázquez García, M.N.; Melen, G.J.; Entrena Martínez, A.; Cubillo Moreno, I.; García-Castro, J.; Ramírez Orellana, M.; Zapata González, A.G. Mesenchymal Stromal Cells Derived from the Bone Marrow of Acute Lymphoblastic Leukemia Patients Show Altered BMP4 Production: Correlations with the Course of Disease. PLoS ONE 2014, 9, e84496. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Cox, C.V.; Blair, A. A primitive cell origin for B-cell precursor ALL? Stem Cell Rev. 2005, 1, 189–196. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Cancer Statistics Review, 1975–2016—SEER Statistics. Available online: https://seer.cancer.gov/archive/csr/1975_2016/ (accessed on 7 March 2022).
- Levine, R.L. Molecular pathogenesis of AML: Translating insights to the clinic. Best Pract. Res. Clin. Haematol. 2013, 26, 245–248. [Google Scholar] [CrossRef][Green Version]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef]
- Zhang, X.; Rastogi, P.; Shah, B.; Zhang, L.; Zhang, X.; Rastogi, P.; Shah, B.; Zhang, L. B lymphoblastic leukemia/lymphoma: New insights into genetics, molecular aberrations, subclassification and targeted therapy. Oncotarget 2017, 8, 66728–66741. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, a034835. [Google Scholar] [CrossRef]
- van den Berk, L.C.J.; van der Veer, A.; Willemse, M.E.; Theeuwes, M.J.G.A.; Luijendijk, M.W.; Tong, W.H.; van der Sluis, I.M.; Pieters, R.; den Boer, M.L. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 166, 240–249. [Google Scholar] [CrossRef]
- Jung, E.M.; Kwon, O.; Kwon, K.S.; Cho, Y.S.; Rhee, S.K.; Min, J.K.; Oh, D.B. Evidences for correlation between the reduced VCAM-1 expression and hyaluronan synthesis during cellular senescence of human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2011, 404, 463–469. [Google Scholar] [CrossRef]
- Singh, P.; Kacena, M.A.; Orschell, C.M.; Pelus, L.M. Aging-Related Reduced Expression of CXCR4 on Bone Marrow Mesenchymal Stromal Cells Contributes to Hematopoietic Stem and Progenitor Cell Defects. Stem Cell Rev. Rep. 2020, 16, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Geißler, S.; Textor, M.; Kühnisch, J.; Könnig, D.; Klein, O.; Ode, A.; Pfitzner, T.; Adjaye, J.; Kasper, G.; Duda, G.N. Functional Comparison of Chronological and In Vitro Aging: Differential Role of the Cytoskeleton and Mitochondria in Mesenchymal Stromal Cells. PLoS ONE 2012, 7, e52700. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, W.J.C.; Ploemacher, R.E. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003, 17, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Bustos, M.L.; Huleihel, L.; Kapetanaki, M.G.; Lino-Cardenas, C.L.; Mroz, L.; Ellis, B.M.; McVerry, B.J.; Richards, T.J.; Kaminski, N.; Cerdenes, N.; et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am. J. Respir. Crit. Care Med. 2014, 189, 787–798. [Google Scholar] [CrossRef]
- Turinetto, V.; Vitale, E.; Giachino, C. Senescence in Human Mesenchymal Stem Cells: Functional Changes and Implications in Stem Cell-Based Therapy. Int. J. Mol. Sci. 2016, 17, 1164. [Google Scholar] [CrossRef]
- Sethe, S.; Scutt, A.; Stolzing, A. Aging of mesenchymal stem cells. Ageing Res. Rev. 2006, 5, 91–116. [Google Scholar] [CrossRef]
- Kasper, G.; Mao, L.; Geissler, S.; Draycheva, A.; Trippens, J.; Kühnisch, J.; Tschirschmann, M.; Kaspar, K.; Perka, C.; Duda, G.N.; et al. Insights into Mesenchymal Stem Cell Aging: Involvement of Antioxidant Defense and Actin Cytoskeleton. Stem Cells 2009, 27, 1288–1297. [Google Scholar] [CrossRef]
- Liu, M.; Lei, H.; Dong, P.; Fu, X.; Yang, Z.; Yang, Y.; Ma, J.; Liu, X.; Cao, Y.; Xiao, R. Adipose-Derived Mesenchymal Stem Cells from the Elderly Exhibit Decreased Migration and Differentiation Abilities with Senescent Properties. Cell Transplant. 2017, 26, 1505–1519. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Burska, A.N.; Ponchel, F.; Giannoudis, P.V.; Jones, E.A. The Analysis of in Vivo Aging in Human Bone Marrow Mesenchymal Stromal Cells Using Colony-Forming Unit-Fibroblast Assay and the CD45lowCD271+ Phenotype. Stem Cells Int. 2019, 2019, 5197983. [Google Scholar] [CrossRef]
- Kapetanos, K.; Asimakopoulos, D.; Christodoulou, N.; Vogt, A.; Khan, W. Chronological Age Affects MSC Senescence In Vitro—A Systematic Review. Int. J. Mol. Sci. 2021, 22, 7945. [Google Scholar] [CrossRef] [PubMed]
- Bonab, M.M.; Alimoghaddam, K.; Talebian, F.; Ghaffari, S.H.; Ghavamzadeh, A.; Nikbin, B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative Senescence of Mesenchymal Stem Cells: A Continuous and Organized Process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Integration of clearance mechanisms: The proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2010, 2, a006734. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hong, Y.; Zhang, H.; Li, X. Mesenchymal Stem Cell Senescence and Rejuvenation: Current Status and Challenges. Front. Cell Dev. Biol. 2020, 8, 364. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Petropoulos, I.; Franceschi, C.; Friguet, B.; Gonos, E.S. Fibroblast cultures from healthy centenarians have an active proteasome. Exp. Gerontol. 2000, 35, 721–728. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Stratford, F.L.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central Role of the Proteasome in Senescence and Survival of Human Fibroblasts: Induction of a Senescence-like Phenotype upon Its Inhibition and Resistance to Stress upon Its Activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef]
- Gaczynska, M.; Osmulskil, P.A.; Ward, W.F. Caretaker or undertaker? The role of the proteasome in aging. Mech. Ageing Dev. 2001, 122, 235–254. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. Aging as an Event of Proteostasis Collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef]
- Capasso, S.; Alessio, N.; Squillaro, T.; Di Bernardo, G.; Melone, M.A.; Cipollaro, M.; Peluso, G.; Galderisi, U.; Capasso, S.; Alessio, N.; et al. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget 2015, 6, 39457–39468. [Google Scholar] [CrossRef]
- Lu, L.; Song, H.F.; Zhang, W.G.; Liu, X.Q.; Zhu, Q.; Cheng, X.L.; Yang, G.J.; Li, A.; Xiao, Z.C. Potential role of 20S proteasome in maintaining stem cell integrity of human bone marrow stromal cells in prolonged culture expansion. Biochem. Biophys. Res. Commun. 2012, 422, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kapetanou, M.; Chondrogianni, N.; Petrakis, S.; Koliakos, G.; Gonos, E.S. Proteasome activation enhances stemness and lifespan of human mesenchymal stem cells. Free Radic. Biol. Med. 2017, 103, 226–235. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912. [Google Scholar] [CrossRef] [PubMed]
- Jakovljevic, J.; Harrell, C.R.; Fellabaum, C.; Arsenijevic, A.; Jovicic, N.; Volarevic, V. Modulation of autophagy as new approach in mesenchymal stem cell-based therapy. Biomed. Pharmacother. 2018, 104, 404–410. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Wang, L.; Han, X.; Qu, G.; Su, L.; Zhao, B.; Miao, J. A pH probe inhibits senescence in mesenchymal stem cells 06 Biological Sciences 0601 Biochemistry and Cell Biology. Stem Cell Res. Ther. 2018, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Hsu, M.F.; Wu, K.K. High Glucose Induces Bone Marrow-Derived Mesenchymal Stem Cell Senescence by Upregulating Autophagy. PLoS ONE 2015, 10, e0126537. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hu, C.J.; Zhuo, R.H.; Lei, Y.S.; Han, N.N.; He, L. Inhibition of autophagy alleviates the senescent state of rat mesenchymal stem cells during long-term culture. Mol. Med. Rep. 2014, 10, 3003–3008. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lei, Y.; Hu, C.; Hu, C. p53 regulates autophagic activity in senescent rat mesenchymal stromal cells. Exp. Gerontol. 2016, 75, 64–71. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol. Cells 2017, 40, 607. [Google Scholar]
- Rastaldo, R.; Vitale, E.; Giachino, C. Dual Role of Autophagy in Regulation of Mesenchymal Stem Cell Senescence. Front. Cell Dev. Biol. 2020, 8, 276. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Ruvolo, P.P.; Wang, R.Y.; Battula, V.L.; Shpall, E.J.; Ruvolo, V.R.; McQueen, T.; Qui, Y.H.; Zeng, Z.; Pierce, S.; et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica 2018, 103, 810–821. [Google Scholar] [CrossRef]
- Zhao, Z.G.; Liang, Y.; Li, K.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Phenotypic and functional comparison of mesenchymal stem cells derived from the bone marrow of normal adults and patients with hematologic malignant diseases. Stem Cells Dev. 2007, 16, 637–648. [Google Scholar] [CrossRef]
- Kim, Y.; Jekarl, D.W.; Kim, J.; Kwon, A.; Choi, H.; Lee, S.; Kim, Y.J.; Kim, H.J.; Kim, Y.; Oh, I.H.; et al. Genetic and epigenetic alterations of bone marrow stromal cells in myelodysplastic syndrome and acute myeloid leukemia patients. Stem Cell Res. 2015, 14, 177–184. [Google Scholar] [CrossRef]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells from De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Becker, P.S. Dependence of acute myeloid leukemia on adhesion within the bone marrow microenvironment. Sci. World J. 2012, 2012, 856467. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Goodarzi, A.; Valikhani, M.; Amiri, F.; Safari, A. The mechanisms of mutual relationship between malignant hematologic cells and mesenchymal stem cells: Does it contradict the nursing role of mesenchymal stem cells? Cell Commun. Signal. 2022, 20, 21. [Google Scholar] [CrossRef]
- Burger, J.A.; Bürkle, A. The CXCR4 chemokine receptor in acute and chronic leukaemia: A marrow homing receptor and potential therapeutic target. Br. J. Haematol. 2007, 137, 288–296. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Forte, D.; Cavo, M.; Curti, A. Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance? Cancers 2021, 13, 5319. [Google Scholar] [CrossRef]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef]
- Yehudai-Resheff, S.; Attias-Turgeman, S.; Sabbah, R.; Gabay, T.; Musallam, R.; Fridman-Dror, A.; Zuckerman, T. Abnormal morphological and functional nature of bone marrow stromal cells provides preferential support for survival of acute myeloid leukemia cells. Int. J. Cancer 2019, 144, 2279–2289. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, e90036. [Google Scholar] [CrossRef]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Y.; Cheng, J.; Ma, J.; Li, Q.; Pang, T. ATG5 regulates mesenchymal stem cells differentiation and mediates chemosensitivity in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2020, 525, 398–405. [Google Scholar] [CrossRef]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef]
- Bonilla, X.; Vanegas, N.D.P.; Vernot, J.P. Acute leukemia induces senescence and impaired osteogenic differentiation in mesenchymal stem cells endowing leukemic cells with functional advantages. Stem Cells Int. 2019, 2019, 3864948. [Google Scholar] [CrossRef]
- Vanegas, N.-D.P.; Fernanda Ruiz-Aparicio, P.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.-P.; Vanegas, N.-D.P.; Ruiz-Aparicio, P.F.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.-P.; et al. Leukemia-Induced Cellular Senescence and Stemness Alterations in Mesenchymal Stem Cells Are Reversible upon Withdrawal of B-Cell Acute Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 8166. [Google Scholar] [CrossRef]
- Ruiz-Aparicio, P.F.; Vanegas, N.D.P.; Uribe, G.I.; Ortiz-Montero, P.; Cadavid-Cortés, C.; Lagos, J.; Flechas-Afanador, J.; Linares-Ballesteros, A.; Vernot, J.P. Dual targeting of stromal cell support and leukemic cell growth by a peptidic pkc inhibitor shows effectiveness against b-all. Int. J. Mol. Sci. 2020, 21, 3705. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef]
- Dimitriou, H.; Linardakis, E.; Martimianaki, G.; Stiakaki, E.; Perdikogianni, C.; Charbord, P.; Kalmanti, M. Properties and potential of bone marrow mesenchymal stromal cells from children with hematologic diseases. Cytotherapy 2008, 10, 125–133. [Google Scholar] [CrossRef]
- Åhsberg, J.; Xiao, P.; Okuyama, K.; Somasundaram, R.; Strid, T.; Qian, H.; Sigvardsson, M. Progression of progenitor B-cell leukemia is associated with alterations of the bone marrow micro-environment. Haematologica 2020, 105, e102–e106. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, B.; Polak, R.; Stalpers, F.; Pieters, R.; Den Boer, M.L. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia 2017, 31, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Valle-Prieto, A.; Conget, P.A. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 2010, 19, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, S.; Tsoyi, K.; Liu, X.; Nakahira, K.; Ith, B.; Coronata, A.A.; Fredenburgh, L.E.; Englert, J.A.; Piantadosi, C.A.; Choi, A.M.K.; et al. Mesenchymal Stromal Cells Deficient in Autophagy Proteins Are Susceptible to Oxidative Injury and Mitochondrial Dysfunction. Am. J. Respir. Cell Mol. Biol. 2017, 56, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef]
- Stab, B.R.; Martinez, L.; Grismaldo, A.; Lerma, A.; Gutiérrez, M.L.; Barrera, L.A.; Sutachan, J.J.; Albarracín, S.L. Mitochondrial functional changes characterization in young and senescent human adipose derived MSCs. Front. Aging Neurosci. 2016, 8, 299. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, L.; Han, S.; Chen, L.; Li, C.; Zhang, Z.; Hong, Y.; Zhang, X.; Zhou, X.; Jiang, D.; et al. Adult mesenchymal stem cell ageing interplays with depressed mitochondrial Ndufs6. Cell Death Dis. 2020, 11, 1075. [Google Scholar] [CrossRef]
- Lonergan, T.; Brenner, C.; Bavister, B. Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J. Cell. Physiol. 2006, 208, 149–153. [Google Scholar] [CrossRef]
- Barilani, M.; Lovejoy, C.; Piras, R.; Abramov, A.Y.; Lazzari, L.; Angelova, P.R. Age-related changes in the energy of human mesenchymal stem cells. J. Cell. Physiol. 2021, 237, 1753–1767. [Google Scholar] [CrossRef]
- Fernandez-Rebollo, E.; Franzen, J.; Goetzke, R.; Hollmann, J.; Ostrowska, A.; Oliverio, M.; Sieben, T.; Rath, B.; Kornfeld, J.W.; Wagner, W. Senescence-Associated Metabolomic Phenotype in Primary and iPSC-Derived Mesenchymal Stromal Cells. Stem Cell Rep. 2020, 14, 201–209. [Google Scholar] [CrossRef]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Bernard, D.; Gil, J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol. Histopathol. 2007, 22, 85–90. [Google Scholar]
- Chondrogianni, N.; Gonos, E.S. Proteasome inhibition induces a senescence-like phenotype in primary human fibroblasts cultures. Biogerontology 2004, 5, 55–61. [Google Scholar] [CrossRef]
- Hladik, D.; Höfig, I.; Oestreicher, U.; Beckers, J.; Matjanovski, M.; Bao, X.; Scherthan, H.; Atkinson, M.J.; Rosemann, M. Long-term culture of mesenchymal stem cells impairs ATM-dependent recognition of DNA breaks and increases genetic instability. Stem Cell Res. Ther. 2019, 10, 218. [Google Scholar] [CrossRef]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef]
- Massaro, F.; Corrillon, F.; Stamatopoulos, B.; Meuleman, N.; Lagneaux, L.; Bron, D. Aging of Bone Marrow Mesenchymal Stromal Cells: Hematopoiesis Disturbances and Potential Role in the Development of Hematologic Cancers. Cancers 2020, 13, 68. [Google Scholar] [CrossRef]
- Whitehead, J.; Zhang, J.; Harvestine, J.N.; Kothambawala, A.; Liu, G.Y.; Leach, J.K. Tunneling nanotubes mediate the expression of senescence markers in mesenchymal stem/stromal cell spheroids. Stem Cells 2020, 38, 80–89. [Google Scholar] [CrossRef]
- Jiang, D.; Gao, F.; Zhang, Y.; Wong, D.S.H.; Li, Q.; Tse, H.F.; Xu, G.; Yu, Z.; Lian, Q. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 2016, 7, e2467. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Vignon, C.; Debeissat, C.; Bourgeais, J.; Gallay, N.; Kouzi, F.; Anginot, A.; Picou, F.; Guardiola, P.; Ducrocq, E.; Foucault, A.; et al. Involvement of GPx-3 in the Reciprocal Control of Redox Metabolism in the Leukemic Niche. Int. J. Mol. Sci. 2020, 21, 8584. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Zhang, Q.; Yang, H.; Yamatani, K.; Ai, T.; Ruvolo, V.; Baran, N.; Cai, T.; Ma, H.; Jacamo, R.; et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021, 5, 4233–4255. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Forte, D.; García-Fernández, M.; Sánchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martín-Pérez, D.; López, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843.e9. [Google Scholar] [CrossRef]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Dempsey, C.; Chadwick, A.; Harrison, S.; Liu, J.; Di, Y.; McGinn, O.J.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P.; et al. Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood 2016, 128, 453. [Google Scholar] [CrossRef] [PubMed]
- Cominal, J.G.; da Costa Cacemiro, M.; Pinto-Simões, B.; Kolb, H.J.; Malmegrim, K.C.R.; Attié de Castro, F. Emerging Role of Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Pathogenesis of Haematological Malignancies. Stem Cells Int. 2019, 2019, 6854080. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Lim, M.; Pang, Y.; Ma, S.; Hao, S.; Shi, H.; Zheng, Y.; Hua, C.; Gu, X.; Yang, F.; Yuan, W.; et al. Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia 2015, 30, 154–162. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, E.W.; Croteau, D.L.; Bohr, V.A. Heterochromatin: An epigenetic point of view in aging. Exp. Mol. Med. 2020, 52, 1466–1474. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- So, A.Y.; Jung, J.W.; Lee, S.; Kim, H.S.; Kang, K.S. DNA Methyltransferase Controls Stem Cell Aging by Regulating BMI1 and EZH2 through MicroRNAs. PLoS ONE 2011, 6, e19503. [Google Scholar] [CrossRef]
- Fernández, A.F.; Bayón, G.F.; Urdinguio, R.G.; Toraño, E.G.; García, M.G.; Carella, A.; Petrus-Reurer, S.; Ferrero, C.; Martinez-Camblor, P.; Cubillo, I.; et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res. 2015, 25, 27–40. [Google Scholar] [CrossRef]
- Johansson, Å.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef]
- Choi, M.R.; In, Y.H.; Park, J.; Park, T.; Jung, K.H.; Chai, J.C.; Chung, M.K.; Lee, Y.S.; Chai, Y.G. Genome-scale DNA methylation pattern profiling of human bone marrow mesenchymal stem cells in long-term culture. Exp. Mol. Med. 2012, 44, 503–512. [Google Scholar] [CrossRef]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef]
- Toraño, E.G.; Bayón, G.F.; del Real, Á.; Sierra, M.I.; García, M.G.; Carella, A.; Belmonte, T.; Urdinguio, R.G.; Cubillo, I.; García-Castro, J.; et al. Age-associated hydroxymethylation in human bone-marrow mesenchymal stem cells. J. Transl. Med. 2016, 14, 207. [Google Scholar] [CrossRef]
- Schellenberg, A.; Joussen, S.; Moser, K.; Hampe, N.; Hersch, N.; Hemeda, H.; Schnitker, J.; Denecke, B.; Lin, Q.; Pallua, N.; et al. Matrix elasticity, replicative senescence and DNA methylation patterns of mesenchymal stem cells. Biomaterials 2014, 35, 6351–6358. [Google Scholar] [CrossRef]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583. [Google Scholar] [CrossRef]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castaño, J.; Gómez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573. [Google Scholar] [CrossRef]
- Lee, H.R.; Yang, S.J.; Choi, H.K.; Kim, J.A.; Oh, I.H. The Chromatin Remodeling Complex CHD1 Regulates the Primitive State of Mesenchymal Stromal Cells to Control Their Stem Cell Supporting Activity. Stem Cells Dev. 2021, 30, 363–373. [Google Scholar] [CrossRef]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef]
- Wen, L.; Li, J.; Guo, H.; Liu, X.; Zheng, S.; Zhang, D.; Zhu, W.; Qu, J.; Guo, L.; Du, D.; et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Res. 2015, 25, 1250–1264. [Google Scholar] [CrossRef]
- Conforti, A.; Biagini, S.; Del Bufalo, F.; Sirleto, P.; Angioni, A.; Starc, N.; Li Pira, G.; Moretta, F.; Proia, A.; Contoli, B.; et al. Biological, Functional and Genetic Characterization of Bone Marrow-Derived Mesenchymal Stromal Cells from Pediatric Patients Affected by Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e76989. [Google Scholar] [CrossRef]
- Menendez, P.; Catalina, P.; Rodríguez, R.; Melen, G.J.; Bueno, C.; Arriero, M.; García-Sánchez, F.; Lassaletta, A.; García-Sanz, R.; García-Castro, J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009, 206, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci. Adv. 2020, 6, eaba5536. [Google Scholar] [CrossRef] [PubMed]
- Jacamo, R.; Chen, Y.; Wang, Z.; Wencai, M.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aparicio, P.F.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.P. Sensitization to Drug Treatment in Precursor B-Cell Acute Lymphoblastic Leukemia Is Not Achieved by Stromal NF-κB Inhibition of Cell Adhesion but by Stromal PKC-Dependent Inhibition of ABC Transporters Activity. Molecules 2021, 26, 5366. [Google Scholar] [CrossRef]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein Kinase C-β-Dependent Activation of NF-κB in Stromal Cells Is Indispensable for the Survival of Chronic Lymphocytic Leukemia B Cells In Vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef]
- Niedermeier, M.; Hennessy, B.T.; Knight, Z.A.; Henneberg, M.; Hu, J.; Kurtova, A.V.; Wierda, W.G.; Keating, M.J.; Shokat, K.M.; Burger, J.A. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: A novel therapeutic approach. Blood 2009, 113, 5549–5557. [Google Scholar] [CrossRef]
- Jones, C.L.; Gearheart, C.M.; Fosmire, S.; Delgado-Martin, C.; Evensen, N.A.; Bride, K.; Waanders, A.J.; Pais, F.; Wang, J.; Bhatla, T.; et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood 2015, 126, 2202–2212. [Google Scholar] [CrossRef]
- Amigo-Jiménez, I.; Bailón, E.; Aguilera-Montilla, N.; Terol, M.J.; García-Marco, J.A.; García-Pardo, A. Bone marrow stroma-induced resistance of chronic lymphocytic leukemia cells to arsenic trioxide involves Mcl-1 upregulation and is overcome by inhibiting the PI3Kδ or PKCβ signaling pathways. Oncotarget 2015, 6, 44832–44848. [Google Scholar] [CrossRef]
- Feldhahn, N.; Arutyunyan, A.; Stoddart, S.; Zhang, B.; Schmidhuber, S.; Yi, S.-J.; Kim, Y.; Groffen, J.; Heisterkamp, N. Environment-mediated drug resistance in Bcr/Abl-positive acute lymphoblastic leukemia. Oncoimmunology 2012, 1, 618. [Google Scholar] [CrossRef]
- Evangelisti, C.; Cappellini, A.; Oliveira, M.; Fragoso, R.; Barata, J.T.; Bertaina, A.; Locatelli, F.; Simioni, C.; Neri, L.M.; Chiarini, F.; et al. Phosphatidylinositol 3-kinase inhibition potentiates glucocorticoid response in B-cell acute lymphoblastic leukemia. J. Cell. Physiol. 2018, 233, 1796–1811. [Google Scholar] [CrossRef]
- Park, E.; Chen, J.; Moore, A.; Mangolini, M.; Santoro, A.; Boyd, J.R.; Schjerven, H.; Ecker, V.; Buchner, M.; Williamson, J.C.; et al. Stromal cell protein kinase C-β inhibition enhances chemosensitivity in B cell malignancies and overcomes drug resistance. Sci. Transl. Med. 2020, 12, eaax9340. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Ergen, A.V.; Boles, N.C.; Goodell, M.A. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 2012, 119, 2500–2509. [Google Scholar] [CrossRef]
- O’Hagan-Wong, K.; Nadeau, S.; Carrier-Leclerc, A.; Apablaza, F.; Hamdy, R.; Shum-Tim, D.; Rodier, F.; Colmegna, I. Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells’ homeostasis. Oncotarget 2016, 7, 13285–13296. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef]
- Adler, B.J.; Green, D.E.; Pagnotti, G.M.; Chan, M.E.; Rubin, C.T. High Fat Diet Rapidly Suppresses B Lymphopoiesis by Disrupting the Supportive Capacity of the Bone Marrow Niche. PLoS ONE 2014, 9, e90639. [Google Scholar] [CrossRef]
- Boulestreau, J.; Maumus, M.; Rozier, P.; Jorgensen, C.; Noël, D. Mesenchymal Stem Cell Derived Extracellular Vesicles in Aging. Front. Cell Dev. Biol. 2020, 8, 107. [Google Scholar] [CrossRef]
- Zhang, Y.; Ravikumar, M.; Ling, L.; Nurcombe, V.; Cool, S.M. Age-Related Changes in the Inflammatory Status of Human Mesenchymal Stem Cells: Implications for Cell Therapy. Stem Cell Rep. 2021, 16, 694–707. [Google Scholar] [CrossRef]
- Lei, Q.; Liu, T.; Gao, F.; Xie, H.; Sun, L.; Zhao, A.; Ren, W.; Guo, H.; Zhang, L.; Wang, H.; et al. Microvesicles as Potential Biomarkers for the Identification of Senescence in Human Mesenchymal Stem Cells. Theranostics 2017, 7, 2673. [Google Scholar] [CrossRef]
- Fafián-Labora, J.; Morente-López, M.; Sánchez-Dopico, M.J.; Arntz, O.J.; Van De Loo, F.A.J.; De Toro, J.; Arufe, M.C. Influence of mesenchymal stem cell-derived extracellular vesicles in vitro and their role in ageing. Stem Cell Res. Ther. 2020, 11, 13. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- Pievani, A.; Biondi, M.; Tomasoni, C.; Biondi, A.; Serafini, M. Location First: Targeting Acute Myeloid Leukemia Within Its Niche. J. Clin. Med. 2020, 9, 1513. [Google Scholar] [CrossRef]
- Kuett, A.; Rieger, C.; Perathoner, D.; Herold, T.; Wagner, M.; Sironi, S.; Sotlar, K.; Horny, H.P.; Deniffel, C.; Drolle, H.; et al. IL-8 as mediator in the microenvironment-leukaemia network in acute myeloid leukaemia. Sci. Rep. 2015, 5, 18411. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef]
- Cheng, J.; Li, Y.; Liu, S.; Jiang, Y.; Ma, J.; Wan, L.; Li, Q.; Pang, T. CXCL8 derived from mesenchymal stromal cells supports survival and proliferation of acute myeloid leukemia cells through the PI3K/AKT pathway. FASEB J. 2019, 33, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Vernot, J.-P.; Bonilla, X.; Rodriguez-Pardo, V.; Vanegas, N.-D. Phenotypic and Functional Alterations of Hematopoietic Stem and Progenitor Cells in an In Vitro Leukemia-Induced Microenvironment. Int. J. Mol. Sci. 2017, 18, 199. [Google Scholar] [CrossRef]
- de Vasconcellos, J.F.; Laranjeira, A.B.A.; Zanchin, N.I.T.; Otubo, R.; Vaz, T.H.; Cardoso, A.A.; Brandalise, S.R.; Yunes, J.A. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 568–577. [Google Scholar] [CrossRef]
- Polak, R.; De Rooij, B.; Pieters, R.; Den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef]
- Kletsas, D.; Pratsinis, H.; Mariatos, G.; Zacharatos, P.; Gorgoulis, V.G. The Proinflammatory Phenotype of Senescent Cells: The p53-Mediated ICAM-1 Expression. Ann. N. Y. Acad. Sci. 2004, 1019, 330–332. [Google Scholar] [CrossRef]
- Giordano, P.; Molinari, A.C.; Del Vecchio, G.C.; Saracco, P.; Russo, G.; Altomare, M.; Perutelli, P.; Crescenzio, N.; Santoro, N.; Marchetti, M.; et al. Prospective study of hemostatic alterations in children with acute lymphoblastic leukemia. Am. J. Hematol. 2010, 85, 325–330. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef]
- Hermann, A.; List, C.; Habisch, H.J.; Vukicevic, V.; Ehrhart-Bornstein, M.; Brenner, R.; Bernstein, P.; Fickert, S.; Storch, A. Age-dependent neuroectodermal differentiation capacity of human mesenchymal stromal cells: Limitations for autologous cell replacement strategies. Cytotherapy 2010, 12, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Yannarelli, G.; Pacienza, N.; Cuniberti, L.; Medin, J.; Davies, J.; Keating, A. Brief Report: The Potential Role of Epigenetics on Multipotent Cell Differentiation Capacity of Mesenchymal Stromal Cells. Stem Cells 2013, 31, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cãamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Cakouros, D.D.; Gronthos, S. Epigenetic Regulation of Bone Marrow Stem Cell Aging: Revealing Epigenetic Signatures associated with Hematopoietic and Mesenchymal Stem Cell Aging. Aging Dis. 2019, 10, 174–189. [Google Scholar] [CrossRef]
- Lu, Y.; Qu, H.; Qi, D.; Xu, W.; Liu, S.; Jin, X.; Song, P.; Guo, Y.; Jia, Y.; Wang, X.; et al. OCT4 maintains self-renewal and reverses senescence in human hair follicle mesenchymal stem cells through the downregulation of p21 by DNA methyltransferases. Stem Cell Res. Ther. 2019, 10, 28. [Google Scholar] [CrossRef]
- Davis, C.; Dukes, A.; Drewry, M.; Helwa, I.; Johnson, M.H.; Isales, C.M.; Hill, W.D.; Liu, Y.; Shi, X.; Fulzele, S.; et al. MicroRNA-183-5p Increases with Age in Bone-Derived Extracellular Vesicles, Suppresses Bone Marrow Stromal (Stem) Cell Proliferation, and Induces Stem Cell Senescence. Tissue Eng. Part A 2017, 23, 1231–1240. [Google Scholar] [CrossRef]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [PubMed]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2019, 34, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.C.; Tickner, J.; Hughes, A.M.; Skut, P.; Howlett, M.; Foley, B.; Oommen, J.; Wells, J.E.; He, B.; Singh, S.; et al. New therapeutic opportunities from dissecting the pre-B leukemia bone marrow microenvironment. Leukemia 2018, 32, 2326–2338. [Google Scholar] [PubMed]
- Rajakumar, S.A.; Papp, E.; Lee, K.K.; Grandal, I.; Merico, D.; Liu, C.C.; Allo, B.; Zhang, L.; Grynpas, M.D.; Minden, M.D.; et al. B cell acute lymphoblastic leukemia cells mediate RANK-RANKL-dependent bone destruction. Sci. Transl. Med. 2020, 12, eaba5942. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Anand, T.; Bhattacharyya, J.; Sharma, A.; Jaganathan, B.G. K562 chronic myeloid leukemia cells modify osteogenic differentiation and gene expression of bone marrow stromal cells. J. Cell Commun. Signal. 2018, 12, 441–450. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Purton, L.E.; Carlin, L.M.; Celso, C. Lo Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Stem Cell 2018, 22, 64–77.e6. [Google Scholar]
- Bolandi, S.M.; Pakjoo, M.; Beigi, P.; Kiani, M.; Allahgholipour, A.; Goudarzi, N.; Khorashad, J.S.; Eiring, A.M. A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia. Cells 2021, 10, 2833. [Google Scholar] [CrossRef]
- Karjalainen, R.; Pemovska, T.; Popa, M.; Liu, M.; Javarappa, K.K.; Majumder, M.M.; Yadav, B.; Tamborero, D.; Tang, J.; Bychkov, D.; et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell–induced protection of AML. Blood 2017, 130, 789–802. [Google Scholar]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, H.; Duan, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci. Rep. 2015, 5, 16228. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.S.; Kim, H.J.; Konopleva, M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Erbani, J.; Tay, J.; Barbier, V.; Levesque, J.P.; Winkler, I.G. Acute Myeloid Leukemia Chemo-Resistance Is Mediated by E-selectin Receptor CD162 in Bone Marrow Niches. Front. Cell Dev. Biol. 2020, 8, 668. [Google Scholar] [CrossRef] [PubMed]
- Boutin, L.; Arnautou, P.; Trignol, A.; Ségot, A.; Farge, T.; Desterke, C.; Soave, S.; Clay, D.; Raffoux, E.; Sarry, J.E.; et al. Mesenchymal stromal cells confer chemoresistance to myeloid leukemia blasts through Side Population functionality and ABC transporter activation. Haematologica 2020, 105, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Churchman, S.M.; Ponchel, F.; Boxall, S.A.; Cuthbert, R.; Kouroupis, D.; Roshdy, T.; Giannoudis, P.V.; Emery, P.; McGonagle, D.; Jones, E.A. Transcriptional profile of native CD271+ multipotential stromal cells: Evidence for multiple fates, with prominent osteogenic and Wnt pathway signaling activity. Arthritis Rheum. 2012, 64, 2632–2643. [Google Scholar] [CrossRef]
- Taniguchi Ishikawa, E.; Gonzalez-Nieto, D.; Ghiaur, G.; Dunn, S.K.; Ficker, A.M.; Murali, B.; Madhu, M.; Gutstein, D.E.; Fishman, G.I.; Barrio, L.C.; et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9071–9076. [Google Scholar] [CrossRef]
- Singh, A.K.; Cancelas, J.A. Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. Int. J. Mol. Sci. 2020, 21, 796. [Google Scholar] [CrossRef]
- Kouzi, F.; Zibara, K.; Bourgeais, J.; Picou, F.; Gallay, N.; Brossaud, J.; Dakik, H.; Roux, B.; Hamard, S.; Le Nail, L.R.; et al. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 2019, 39, 1198–1212. [Google Scholar] [CrossRef]
- Lopes, M.R.; Pereira, J.K.N.; De Melo Campos, P.; Machado-Neto, J.A.; Traina, F.; Saad, S.T.O.; Favaro, P. De novo AML exhibits greater microenvironment dysregulation compared to AML with myelodysplasia-related changes. Sci. Rep. 2017, 7, 40707. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Bergua, J.M.; Campos, C.; Gayoso, I.; Arcos, M.J.; Bañas, H.; Morgado, S.; Casado, J.G.; Solana, R.; Tarazona, R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: Survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 2013, 61, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, S.; Sun, K.; Chng, W.-J.; Zhou, J.; Wang, S.; Sun, K.; Chng, W.-J. The emerging roles of exosomes in leukemogeneis. Oncotarget 2016, 7, 50698–50707. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Jeong, E.; Boyiadzis, M.; Whiteside, T.L. Increased small extracellular vesicle secretion after chemotherapy via upregulation of cholesterol metabolism in acute myeloid leukaemia. J. Extracell. Vesicles 2020, 9, 1800979. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, G.; Kong, L.; Xu, S.; Wang, Y.; Dong, M. Leukemia-derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci. 2019, 221, 187–195. [Google Scholar] [CrossRef]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribiero, R.; Edwards V, D.K.; Agarwal, A.; Lopez, C.; Jorgens, D.; Tyner, J.W.; et al. Fgf2-fgfr1 signaling regulates release of leukemia-protective exosomes from bone marrow stromal cells. eLife 2019, 8, e40033. [Google Scholar] [CrossRef]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I–II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef]
- Roboz, G.J.; Ritchie, E.K.; Dault, Y.; Lam, L.; Marshall, D.C.; Cruz, N.M.; Hsu, H.T.C.; Hassane, D.C.; Christos, P.J.; Ippoliti, C.; et al. Phase I trial of plerixafor combined with decitabine in newly diagnosed older patients with acute myeloid leukemia. Haematologica 2018, 103, 1308. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S.; Cortes, J.E.; Tse, S.; Gasior, Y.; Jain, N.; Jabbour, E.J.; Estrov, Z.; Alvarado, Y.; DiNardo, C.D.; et al. A phase 1/2 study of ruxolitinib and decitabine in patients with post-myeloproliferative neoplasm acute myeloid leukemia. Leukemia 2020, 34, 2489–2492. [Google Scholar] [CrossRef]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef]
- Randhawa, S.; Cho, B.S.; Ghosh, D.; Sivina, M.; Koehrer, S.; Müschen, M.; Peled, A.; Davis, R.E.; Konopleva, M.; Burger, J.A. Effects of pharmacological and genetic disruption of CXCR4 chemokine receptor function in B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Moses, B.S.; Slone, W.L.; Thomas, P.; Evans, R.; Piktel, D.; Angel, P.M.; Walsh, C.M.; Cantrell, P.S.; Rellick, S.L.; Martin, K.H.; et al. Bone marrow microenvironment modulation of acute lymphoblastic leukemia phenotype. Exp. Hematol. 2016, 44, 50–59.e2. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Jin, L.; Tsutsumi-Ishii, Y.; Xu, Y.; McQueen, T.; Priebe, W.; Mills, G.B.; Ohsaka, A.; Nagaoka, I.; Andreeff, M.; et al. Activation of Integrin-Linked Kinase Is a Critical Prosurvival Pathway Induced in Leukemic Cells by Bone Marrow-Derived Stromal Cells. Cancer Res. 2007, 67, 684–694. [Google Scholar] [CrossRef]
- Ma, Z.; Zhao, X.; Deng, M.; Huang, Z.; Wang, J.; Wu, Y.; Cui, D.; Liu, Y.; Liu, R.; Ouyang, G. Bone Marrow Mesenchymal Stromal Cell-Derived Periostin Promotes B-ALL Progression by Modulating CCL2 in Leukemia Cells. Cell Rep. 2019, 26, 1533–1543.e4. [Google Scholar] [CrossRef]
- Cheung, W.C.; Van Ness, B. The bone marrow stromal microenvironment influences myeloma therapeutic response in vitro. Leukemia 2001, 15, 264–271. [Google Scholar] [CrossRef]
- Kamdje, A.H.N.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Yang, G.C.; Xu, Y.H.; Chen, H.X.; Wang, X.J. Acute Lymphoblastic Leukemia Cells Inhibit the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts in Vitro by Activating Notch Signaling. Stem Cells Int. 2015, 2015, 162410. [Google Scholar] [CrossRef]
- Khan, N.I.; Bradstock, K.F.; Bendall, L.J. Activation of Wnt/β-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia. Br. J. Haematol. 2007, 138, 338–348. [Google Scholar] [CrossRef]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.B.; Lee, J.S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef]
- Sison, E.A.R.; Magoon, D.; Li, L.; Annesley, C.E.; Small, D.; Brown, P. Plerixafor as a chemosensitizing agent in pediatric acute lymphoblastic leukemia: Efficacy and potential mechanisms of resistance to CXCR4 inhibition. Oncotarget 2014, 5, 8947–8958. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, R.; Yu, M.; Lim, M.; Groffen, J.; Heisterkamp, N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia 2011, 25, 1314–1323. [Google Scholar] [CrossRef]
- Schneider, P.; Vasse, M.; Al Bayati, A.; Lenormand, B.; Vannier, J.P. Is high expression of the chemokine receptor CXCR-4 of predictive value for early relapse in childhood acute lymphoblastic leukaemia? Br. J. Haematol. 2002, 119, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.W.; Shi, J.; Chen, J.; Wang, B.; Yu, Y.H.; Qin, X.; Zhou, X.C.; Cai, Y.J.; Li, Z.Q.; Zhang, F.; et al. Leukemia Propagating Cells Rebuild an Evolving Niche in Response to Therapy. Cancer Cell 2014, 25, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Taurino, G.; Dander, E.; Bardelli, D.; Fallati, A.; Andreoli, R.; Bianchi, M.G.; Carubbi, C.; Pozzi, G.; Galuppo, L.; et al. ALL blasts drive primary mesenchymal stromal cells to increase asparagine availability during asparaginase treatment. Blood Adv. 2021, 5, 5164–5178. [Google Scholar] [CrossRef]
- Liu, J.; Masurekar, A.; Johnson, S.; Chakraborty, S.; Griffiths, J.; Smith, D.; Alexander, S.; Dempsey, C.; Parker, C.; Harrison, S.; et al. Stromal cell-mediated mitochondrial redox adaptation regulates drug resistance in childhood acute lymphoblastic leukemia. Oncotarget 2015, 6, 43048–43064. [Google Scholar] [CrossRef][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Aparicio, P.F.; Vernot, J.-P. Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities. J. Pers. Med. 2022, 12, 716. https://doi.org/10.3390/jpm12050716
Ruiz-Aparicio PF, Vernot J-P. Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities. Journal of Personalized Medicine. 2022; 12(5):716. https://doi.org/10.3390/jpm12050716
Chicago/Turabian StyleRuiz-Aparicio, Paola Fernanda, and Jean-Paul Vernot. 2022. "Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities" Journal of Personalized Medicine 12, no. 5: 716. https://doi.org/10.3390/jpm12050716
APA StyleRuiz-Aparicio, P. F., & Vernot, J.-P. (2022). Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities. Journal of Personalized Medicine, 12(5), 716. https://doi.org/10.3390/jpm12050716