
Genetic and Phenotypic Spectrum of KBG Syndrome: A Report of 13 New Chinese Cases and a Review of the Literature

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Genetic Test Methods
2.3. Literature Review
2.4. Statistical Analysis
3. Results
3.1. Clinical Data of the 13 Patients in This Cohort
3.2. Genetic Analysis of the 13 Patients in This Study
3.3. Clinical Characteristics of Reported KBGS Patients
3.4. Summary of the Phenotypic Characteristics of the Patients with Missense Variation
4. Discussion
4.1. Prevalence of KBGS
4.2. Clinical Malformation of KBGS
4.3. Relationship between Genotype and Phenotype
4.4. Clinical Therapy of KBGS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, J.; Pallister, P.D.; Tiddy, W.; Opitz, J.M. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Artic. Ser. 1975, 11, 7–18. [Google Scholar]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 Cause KBG Syndrome, Characterized by Intellectual Disability, Skeletal Malformations, and Macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Li, C.-W.; Chen, J.D. Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem. Biophys. Res. Commun. 2007, 358, 1034–1040. [Google Scholar] [CrossRef] [Green Version]
- Barbaric, I.; Perry, M.J.; Dear, T.N.; Da Costa, A.R.; Salopek, D.; Marusic, A.; Hough, T.; Wells, S.; Hunter, A.J.; Cheeseman, M.; et al. An ENU-induced mutation in the Ankrd11 gene results in an osteopenia-like phenotype in the mouse mutant Yoda. Physiol. Genom. 2008, 32, 311–321. [Google Scholar] [CrossRef]
- Walz, K.; Cohen, D.; Neilsen, P.M.; Foster, J., II; Brancati, F.; Demir, K.; Fisher, R.; Moffat, M.; Verbeek, N.E.; Bjorgo, K.; et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum. Genet. 2015, 134, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Ka, M.; Kim, W.Y. ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol. Dis. 2018, 111, 138–152. [Google Scholar] [CrossRef]
- Gallagher, D.; Voronova, A.; Zander, M.A.; Cancino, G.I.; Bramall, A.; Krause, M.P.; Abad, C.; Tekin, M.; Neilsen, P.M.; Callen, D.F.; et al. Ankrd11 Is a Chromatin Regulator Involved in Autism that Is Essential for Neural Development. Dev. Cell 2015, 32, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, M.; Stein, J.; Grau, L.; Nelson, V.; Meck, J.; Aradhya, S.; Duby, J. Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am. J. Med. Genet. A 2013, 161, 835–840. [Google Scholar] [CrossRef]
- Swols, D.M.; Foster, J., II; Tekin, M. KBG syndrome. Orphanet J. Rare Dis. 2017, 12, 183. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Isrie, M.; Hendriks, Y.; Gielissen, N.; Sistermans, E.A.; Willemsen, M.H.; Peeters, H.; Vermeesch, J.R.; Kleefstra, T.; Van Esch, H. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Europ. J. Hum. Genet. 2012, 20, 131–133. [Google Scholar] [CrossRef]
- Xu, M.; Zhou, H.; Yong, J.; Cong, P.; Li, C.; Yu, Y.; Qi, M. A Chinese patient with KBG syndrome and a 9q31.2-33.1 microdeletion. Eur. J. Med. Genet. 2013, 56, 245–250. [Google Scholar] [CrossRef]
- Tunovic, S.; Barkovich, J.; Sherr, E.H.; Slavotinek, A.M. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am. J. Med. Genet. A 2014, 164, 1744–1749. [Google Scholar] [CrossRef]
- Samanta, D.; Willis, E. Electroencephalographic findings in KBG syndrome: A child with novel mutation in ANKRD11 gene. Acta Neurol. Belg. 2015, 115, 779–782. [Google Scholar] [CrossRef]
- Crippa, M.; Rusconi, D.; Castronovo, C.; Bestetti, I.; Russo, S.; Cereda, A.; Selicorni, A.; Larizza, L.; Finelli, P. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet. 2015, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cho, E.; Park, J.B.; Im, W.Y.; Kim, H.J. A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur. J. Med. Genet. 2015, 58, 86–94. [Google Scholar] [CrossRef]
- Ockeloen, C.W.; Willemsen, M.H.; de Munnik, S.; van Bon, B.W.M.; de Leeuw, N.; Verrips, A.; Kant, S.G.; Jones, E.A.; Brunner, H.G.; van Loon, R.L.E.; et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Europ. J. Hum. Genet. 2015, 23, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Reynaert, N.; Ockeloen, C.W.; Savendahl, L.; Beckers, D.; Devriendt, K.; Kleefstra, T.; Carels, C.E.L.; Grigelioniene, G.; Nordgren, A.; Francois, I.; et al. Short Stature in KBG Syndrome: First Responses to Growth Hormone Treatment. Horm. Res. Paediatr. 2015, 83, 361–364. [Google Scholar] [CrossRef]
- Goldenberg, A.; Riccardi, F.; Tessier, A.; Pfundt, R.; Busa, T.; Cacciagli, P.; Capri, Y.; Coutton, C.; Delahaye-Duriez, A.; Frebourg, T.; et al. Clinical and Molecular Findings in 39 Patients with KBG Syndrome Caused by Deletion or Mutation of ANKRD11. Am. J. Med. Genet. A 2016, 170, 2847–2859. [Google Scholar] [CrossRef]
- Kleyner, R.; Malcolmson, J.; Tegay, D.; Ward, K.; Maughan, A.; Maughan, G.; Nelson, L.; Wang, K.; Robison, R.; Lyon, G.J. KBG syndrome involving a single-nucleotide duplication in ANKRD11. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001131. [Google Scholar] [CrossRef] [Green Version]
- Low, K.; Ashraf, T.; Canham, N.; Clayton-Smith, J.; Deshpande, C.; Donaldson, A.; Fisher, R.; Flinter, F.; Foulds, N.; Fryer, A.; et al. Clinical and Genetic Aspects of KBG Syndrome. Am. J. Med. Genet. A 2016, 170, 2835–2846. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Graul-Neumann, L.; Azzollini, J.; Braunholz, D.; Watrin, E.; Wendt, K.S.; Cereda, A.; Cittaro, D.; et al. Broadening of cohesinopathies: Exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin. Genet. 2016, 89, 74–81. [Google Scholar] [CrossRef]
- Bianchi, P.M.; Bianchi, A.; Digilio, M.C.; Tucci, F.M.; Sitzia, E.; De Vincentiis, G.C. Audiological findings in a de novo mutation of ANKRD11 gene in KBG syndrome: Report of a case and review of the literature. Int. J. Pediatr. Otorhinolaryngol. 2017, 103, 109–112. [Google Scholar] [CrossRef]
- Low, K.J.; Hills, A.; Williams, M.; Duff-Farrier, C.; McKee, S.; Smithson, S.F. A splice-site variant in ANKRD11 associated with classical KBG syndrome. Am. J. Med. Genet. A 2017, 173, 2844–2846. [Google Scholar] [CrossRef]
- Meyer, R.; Soellner, L.; Begemann, M.; Dicks, S.; Fekete, G.; Rahner, N.; Zerres, K.; Elbracht, M.; Eggermann, T. Targeted Next Generation Sequencing Approach in Patients Referred for Silver-Russell Syndrome Testing Increases the Mutation Detection Rate and Provides Decisive Information for Clinical Management. J. Pediatr. 2017, 187, 206–2012.e1. [Google Scholar] [CrossRef]
- Miyatake, S.; Okamoto, N.; Stark, Z.; Nabetani, M.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Mizuguchi, T.; Ohtake, A.; Saitsu, H.; et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J. Hum. Genet. 2017, 62, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Murray, N.; Burgess, B.; Hay, R.; Colley, A.; Rajagopalan, S.; McGaughran, J.; Patel, C.; Enriquez, A.; Goodwin, L.; Stark, Z.; et al. KBG syndrome: An Australian experience. Am. J. Med. Genet. A 2017, 173, 1866–1877. [Google Scholar] [CrossRef]
- Popp, B.; Ekici, A.B.; Thiel, C.T.; Hoyer, J.; Wiesener, A.; Kraus, C.; Reis, A.; Zweier, C. Exome Pool-Seq in neurodevelopmental disorders. Europ. J. Hum. Genet. 2017, 25, 1364–1376. [Google Scholar] [CrossRef] [Green Version]
- De Bernardi, M.L.; Ivanovski, I.; Caraffi, S.G.; Maini, I.; Street, M.E.; Bayat, A.; Zollino, M.; Lepri, F.R.; Gnazzo, M.; Errichiello, E.; et al. Prominent and elongated coccyx, a new manifestation of KBG syndrome associated with novel mutation in ANKRD11. Am. J. Med. Genet. A 2018, 176, 1991–1995. [Google Scholar] [CrossRef]
- Miao, P.; Feng, J.; Guo, Y.; Wang, J.; Xu, X.; Wang, Y.; Li, Y.; Gao, L.; Zheng, C.; Cheng, H. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin. Genet. 2018, 94, 512–520. [Google Scholar] [CrossRef]
- Aitken, S.; Firth, H.V.; McRae, J.; Halachev, M.; Kini, U.; Parker, M.J.; Lees, M.M.; Lachlan, K.; Sarkar, A.; Joss, S.; et al. Finding Diagnostically Useful Patterns in Quantitative Phenotypic Data. Am. J. Hum. Genet. 2019, 105, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, P.; Demaria, F.; Licchelli, S.; Santonastaso, O.; Caciolo, C.; Digilio, M.C.; Sinibaldi, L.; Leoni, C.; Gnazzo, M.; Tartaglia, M.; et al. Obsessive Compulsive Symptoms and Psychopathological Profile in Children and Adolescents with KBG Syndrome. Brain Sci. 2019, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Alves, R.M.; Uva, P.; Veiga, M.F.; Oppo, M.; Zschaber, F.C.R.; Porcu, G.; Porto, H.P.; Persico, I.; Onano, S.; Cuccuru, G.; et al. Novel ANKRD11 gene mutation in an individual with a mild phenotype of KBG syndrome associated to a GEFS plus phenotypic spectrum: A case report. BMC Med. Genet. 2019, 20, 16. [Google Scholar] [CrossRef]
- Aoi, H.; Mizuguchi, T.; Ceroni, J.R.; Kim, V.E.H.; Furquim, I.; Honjo, R.S.; Iwaki, T.; Suzuki, T.; Sekiguchi, F.; Uchiyama, Y.; et al. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J. Hum. Genet. 2019, 64, 967–978. [Google Scholar] [CrossRef]
- Homma, T.K.; Freire, B.L.; Honjo Kawahira, R.S.; Dauber, A.; de Assis Funari, M.F.; Lerario, A.M.; Nishi, M.Y.; de Albuquerque, E.V.; de Andrade Vasques, G.; Collett-Solberg, P.F.; et al. Genetic Disorders in Prenatal Onset Syndromic Short Stature Identified by Exome Sequencing. J. Pediatr. 2019, 215, 192–198. [Google Scholar] [CrossRef]
- Kang, Y.; He, D.; Li, Y.; Zhang, Y.; Shao, Q.; Zhang, M.; Ban, B. A heterozygous point mutation of the ANKRD11 (c.2579C>T) in a Chinese patient with idiopathic short stature. Mol. Genet. Genom. Med. 2019, 7, e988. [Google Scholar] [CrossRef] [Green Version]
- Libianto, R.; Wu, K.H.; Devery, S.; Eisman, J.A.; Center, J.R. KBG syndrome presenting with brachydactyly type E. Bone 2019, 123, 18–22. [Google Scholar] [CrossRef]
- Scarano, E.; Tassone, M.; Graziano, C.; Gibertoni, D.; Tamburrino, F.; Perri, A.; Gnazzo, M.; Severi, G.; Lepri, F.; Mazzanti, L. Novel Mutations and Unreported Clinical Features in KBG Syndrome. Mol. Syndromol. 2019, 10, 130–138. [Google Scholar] [CrossRef]
- Cianci, P.; Pezzoli, L.; Maitz, S.; Agosti, M.; Iascone, M.; Selicorni, A. Dual genetic diagnoses: Neurofibromatosis type 1 and KBG syndrome. Clin. Dysmorphol. 2020, 29, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Cucco, F.; Sarogni, P.; Rossato, S.; Alpa, M.; Patimo, A.; Latorre, A.; Magnani, C.; Puisac, B.; Ramos, F.J.; Pie, J.; et al. Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia de Lange syndrome. Am. J. Med. Genet. A 2020, 182, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-Y.; Ge, L.; Hu, W.-W.; Li, X.-L.; Hu, Y.-Y. Growth hormone therapy for children with KBG syndrome: A case report and review of literature. World J. Clin. Cases 2020, 8, 1172–1179. [Google Scholar] [CrossRef]
- Gnazzo, M.; Lepri, F.R.; Dentici, M.L.; Capolino, R.; Pisaneschi, E.; Agolini, E.; Rinelli, M.; Alesi, V.; Versacci, P.; Genovese, S.; et al. KBG syndrome: Common and uncommon clinical features based on 31 new patients. Am. J. Med. Genet. A 2020, 182, 1073–1083. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Guevara, C.; Guevara, A.; Gavilanes, A.A. Branding of subjects affected with genetic syndromes of severe short stature in developing countries. BMJ Case Rep. 2020, 13, e231737. [Google Scholar] [CrossRef] [Green Version]
- Reuter, M.S.; Chaturvedi, R.R.; Liston, E.; Manshaei, R.; Aul, R.B.; Bowdin, S.; Cohn, I.; Curtis, M.; Dhir, P.; Hayeems, R.Z.; et al. The Cardiac Genome Clinic: Implementing genome sequencing in pediatric heart disease. Genet. Med. 2020, 22, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Sayed, I.S.M.; Abdel-Hamid, M.S.; Abdel-Salam, G.M.H. KBG syndrome in two patients from Egypt. Am. J. Med. Genet. A 2020, 182, 1309–1312. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morisada, N.; Suzuki, T.; Ohashi, Y.; Ye, M.J.; Nozu, K.; Tsuruta, S.; Iijima, K. A woman with a dual genetic diagnosis of autosomal dominant tubulointerstitial kidney disease and KBG syndrome. CEN Case Rep. 2021, 10, 184–188. [Google Scholar] [CrossRef]
- Kim, S.J.; Yang, A.; Park, J.S.; Kwon, D.G.; Lee, J.-S.; Kwon, Y.S.; Lee, J.E. Two Novel Mutations of ANKRD11 Gene and Wide Clinical Spectrum in KBG Syndrome: Case Reports and Literature Review. Front. Genet. 2020, 11, 579805. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, Y.; Yin, X.; Wang, X.; Ni, J.; Dong, Z.; Li, C.; Lu, W. Two loss-of-function ANKRD11 variants in Chinese patients with short stature and a possible molecular pathway. Am. J. Med. Genet. A 2021, 185, 710–718. [Google Scholar] [CrossRef]
- Mattei, D.; Cavarzere, P.; Gaudino, R.; Antoniazzi, F.; Piacentini, G. DYSMORPHIC features and adult short stature: Possible clinical markers of KBG syndrome. Ital. J. Pediatr. 2021, 47, 15. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Mallozzi, M.B.; Huening, I.; Gervasini, C.; Kuechler, A.; Agolini, E.; Albrecht, B.; Baquero-Montoya, C.; Bohring, A.; Bramswig, N.C.; et al. ANKRD11 variants: KBG syndrome and beyond. Clin. Genet. 2021, 100, 187–200. [Google Scholar] [CrossRef]
- Li, Q.; Yang, L.; Wu, J.; Lu, W.; Zhang, M.; Luo, F. A case of KBG syndrome caused by mutation of ANKRD11 gene and literature review. Chin. J. Evid. Based Pediatr. 2018, 13, 452–458. [Google Scholar]
- Wang, D.; Lai, P.; Li, X. Analysis of ANKRD11 gene variant in a family affected with KBG syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi = Zhonghua Yixue Yichuanxue Zazhi = Chin. J. Med. Genet. 2020, 37, 1029–1031. [Google Scholar]
- Cao, Y.; Zhang, L.; Cao, K.; Zhang, G. KBG syndrome: A case report and literature review. J. Chinical Pediatr. 2020, 38, 335–338. [Google Scholar]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.M.; Baddam, P.; Lin, H.; Vidal-Garcia, M.; Aponte, J.D.; De Souza, S.T.; Godziuk, D.; Watson, A.E.S.; Footz, T.; Schachter, N.F.; et al. The Chromatin Regulator Ankrd11 Controls Palate and Cranial Bone Development. Front. Cell Dev. Biol. 2021, 9, 645386. [Google Scholar] [CrossRef]
- Pilon, N. Treatment and Prevention of Neurocristopathies. Trends Mol. Med. 2021, 27, 451–468. [Google Scholar] [CrossRef]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [Green Version]

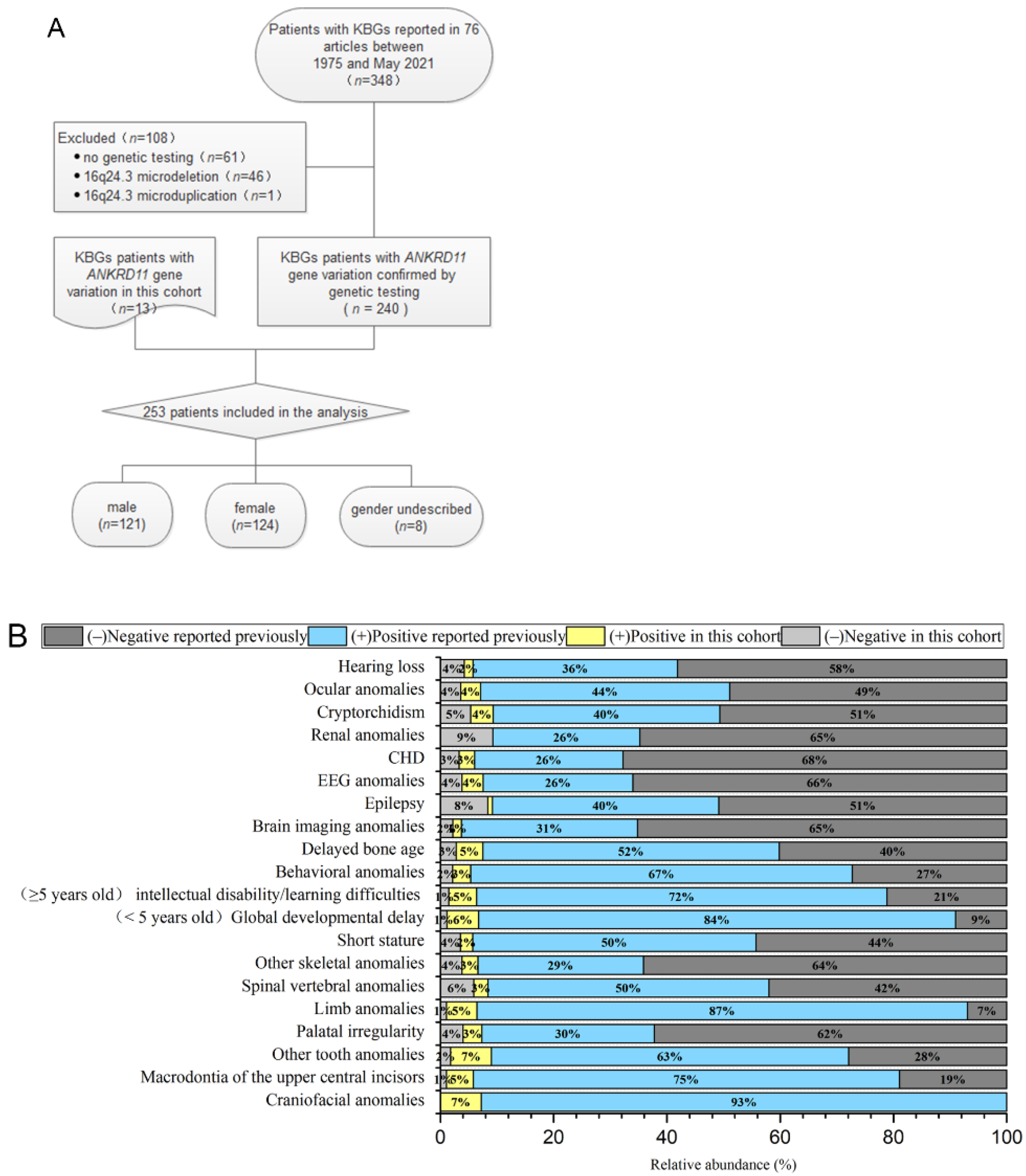
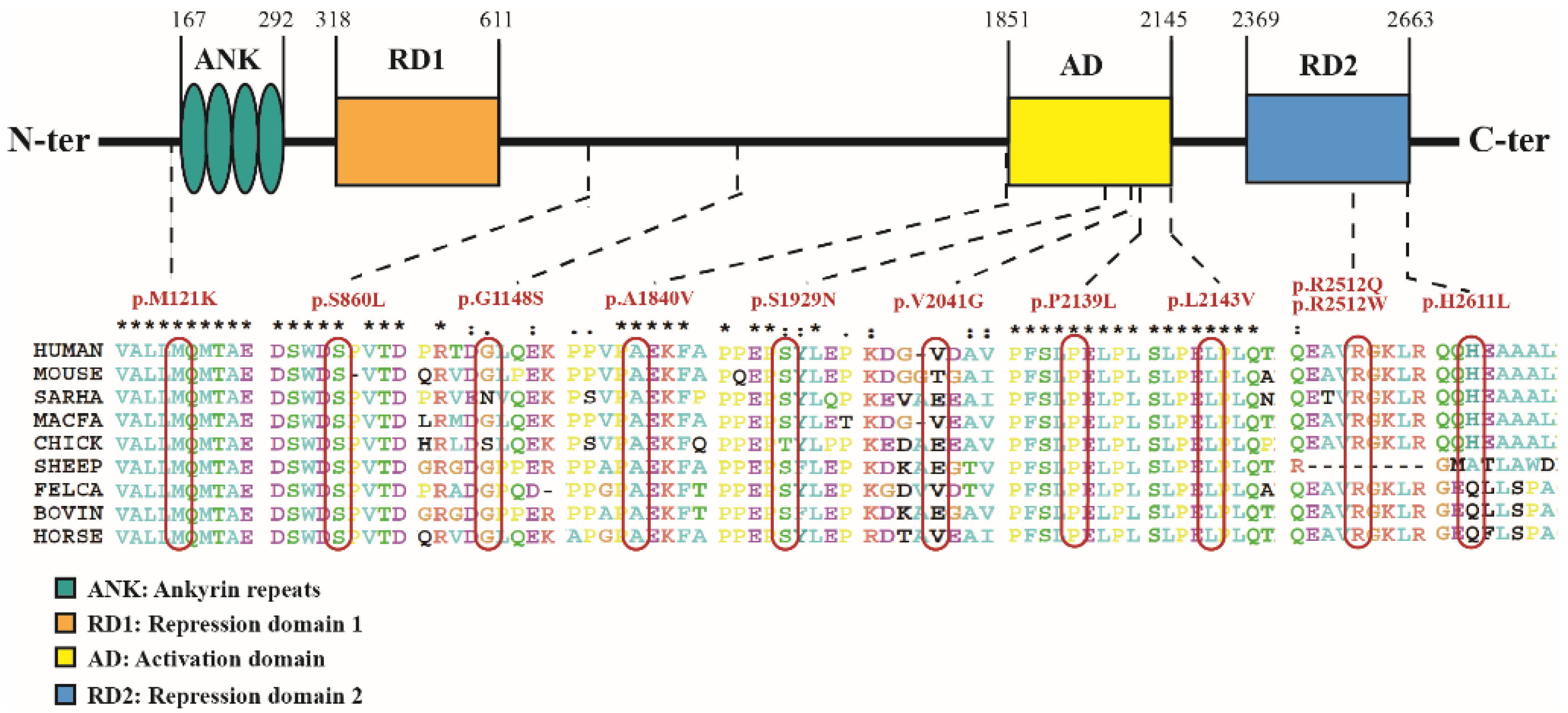

{kind=link}
{kind=link}
{kind=link}
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | P13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | c.3562C > T (p.R1188*) | c.2398_2401del (p.E800Nfs*62) | c.4911delT (p.P1638Lfs*48) | c.5659C > T (p.Q1887*) | c.1801C > T (p.R601*) | c.1903_1907del (p.K635Qfs*26) | c.1903_1907del (p.K635Qfs*26) | c.1903_1907del (p.K635Qfs*26) | c.2262dupA (p.E755Rfs*27) | c.5519C > T (p.A1840V) | c.7832A > T (p.H2611L) | c.6122T > G (p.V2041G) | c.6528_6538del (p.G2177Hfs*5) |
| Variation source | De novo | De novo | De novo | Na | De novo | De novo | Na | De novo | De novo | Mother | De novo | Father | De novo |
| Age at diagnosis | 3y4m | 1y2m | 5y10m | 3y8m | 15y3m | 7m17d | 7y8m | 3y6m | 6y4m | 12y | 3y6m | 4y8m | 7y |
| Gender | Male | Male | Male | Female | Male | Male | Female | Female | Female | Male | Female | Male | Female |
| ACMG | P | P | P | LP | P | P | P | P | P | VUS | LP | VUS | P |
| Birth history | +SGA | − | − | − | − | Na | − | − | − | − | − | +SGA | − |
| Perinatal issues | − | +Feeding difficulties | +Feeding difficulties, vomit | +Feeding difficulties, vomit | +Feeding difficulties | Na | − | +Hypotonia | +Feeding difficulties | − | Na | +Feeding difficulties, large fontanelles | − |
| Craniofacial anomalies | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Dental anomalies | |||||||||||||
| Macrodontia | + | + | + | + | + | Na | + | + | − | + | + | + | Na |
| Other dental anomalies a | + | − | + | + | − | Na | + | + | + | − | Na | + | Na |
| Skeletal anomalies | |||||||||||||
| High palate | + | Na | − | + | − | + | − | − | + | − | + | + | + |
| Clinodactyly of the 5th finger | + | + | − | + | − | Na | + | + | + | − | + | + | + |
| Short finger/small hand | − | − | − | − | + | Na | + | + | + | − | − | − | − |
| scoliosis/kyphosis | − | − | − | Na | Na | Na | Na | − | − | + | − | − | + |
| Other skeletal anomalies b | + | − | + | Na | Na | Na | Na | + | – | Na | – | – | + |
| Short stature | −, 92.2 cm (−1.8 SD) | −, 77 cm (−0.6 SD) | +, 99.8 cm (−3.8 SD) | +, 91 cm (−2.7 SD) | −, 160 cm (−1.6 SD) | −, 69.5 cm (−0.2 SD) | −, 123 cm (−0.7 SD) | −, 99 cm (−0.6 SD) | −, 114 cm (−1.5 SD) | +, 132 cm (−3.2 SD) | +, 92 cm (−2 SD) | +, 101 cm (−2 SD) | −, 116.5 cm (−1.9 SD) |
| Growth retardation | |||||||||||||
| Global development delay | + | + | + | + | − | + | + | + | + | − | Na | + | + |
| Speech and language development delay | +22 mo | + | − | − | − | Na | +4 yr | − | Na | +22 mo | + | − | + |
| Intellectual disability/learning difficulties | + | + | + | − | + | + | + | +IQ79 | − | + | +IQ74 | − | + |
| Behavioural anomalies | − | − | − | +Hyperactivity | Na | Na | +Hyperactivity, short attention span | − | +Short attention span | +Hyperactivity, short attention span | +less communication | +short attention span, timid | Na |
| Delayed bone age | Na | Na | + | + | + | Na | −Advanced | − | + | + | − | Na | Na |
| Neurological abnormalities | |||||||||||||
| Epilepsy | − | − | − | − | Na | − | − | + | − | − | Na | − | − |
| EEG anomalies | Na | + | Na | Na | Na | − | Na | + | Na | Na | Na | − | Na |
| Brain imaging anomalies | Na, pituitary MRI(−) | +Left ventricle slightly wider | − | Na | +Small pituitary | Na | Na, pituitary MRI(−) | - | Na | Na, pituitary MRI(−) | Na | − | Na |
| Ocular anomalies | − | Na | − | − | +Visual field defect | Na | +Strabismus | Na | +Strabismus | − | − | +Astigmatism | +Myopia |
| Hearing loss | − | Na | − | − | + | − | − | − | + | − | Na | +Recurrent otitis media | − |
| CHD | +PDA | − | +VSD | Na | − | +VSD | +VSD | − | Na | − | − | + | − |
| Cryptorchidism | + | − | + | / | − | − | / | / | / | + | / | − | / |
| Renal anomalies | − | − | − | Na | − | Na | Na | Na | Na | Na | Na | − | Na |
| 1st degree relative with KBG | − | − | − | Na | − | − | − | − | − | − | − | − | − |
| Additional features | Deviated nasal septum, sinusitis, preauricular skin tag, supra-auricular pit | Head pilomatrixoma, large fontanelles | Inguinal hernia, micropenis | − | Delayed puberty, micropenis, small testicles, hypogonadotropic hypogonadism, enlarged vestibular aqueduct, obesity | − | Central precocious puberty | Supra-auricular pit | − | – | − | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, F.; Zhao, X.; Cao, B.; Fan, X.; Li, X.; Li, L.; Sui, S.; Su, Z.; Gong, C. Genetic and Phenotypic Spectrum of KBG Syndrome: A Report of 13 New Chinese Cases and a Review of the Literature. J. Pers. Med. 2022, 12, 407. https://doi.org/10.3390/jpm12030407
Gao F, Zhao X, Cao B, Fan X, Li X, Li L, Sui S, Su Z, Gong C. Genetic and Phenotypic Spectrum of KBG Syndrome: A Report of 13 New Chinese Cases and a Review of the Literature. Journal of Personalized Medicine. 2022; 12(3):407. https://doi.org/10.3390/jpm12030407
Chicago/Turabian StyleGao, Fenqi, Xiu Zhao, Bingyan Cao, Xin Fan, Xiaoqiao Li, Lele Li, Shengbin Sui, Zhe Su, and Chunxiu Gong. 2022. "Genetic and Phenotypic Spectrum of KBG Syndrome: A Report of 13 New Chinese Cases and a Review of the Literature" Journal of Personalized Medicine 12, no. 3: 407. https://doi.org/10.3390/jpm12030407
APA StyleGao, F., Zhao, X., Cao, B., Fan, X., Li, X., Li, L., Sui, S., Su, Z., & Gong, C. (2022). Genetic and Phenotypic Spectrum of KBG Syndrome: A Report of 13 New Chinese Cases and a Review of the Literature. Journal of Personalized Medicine, 12(3), 407. https://doi.org/10.3390/jpm12030407