
Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Equipment
2.1. Reagents
2.2. Equipment
2.3. Transfer Plasmid
2.4. Cell Culture Reagents
2.4.1. Human Neuroblastoma Tissue Acquisition
2.4.2. Neuroblastoma Tissue Processing
2.4.3. Neuroblastoma Organoid Medium
2.4.4. DIPG Organoid Medium
2.4.5. Neuroblastoma Cell Culture
2.4.6. DIPG Cell Culture
2.4.7. HEK293 Cell Culture
2.5. Statistics
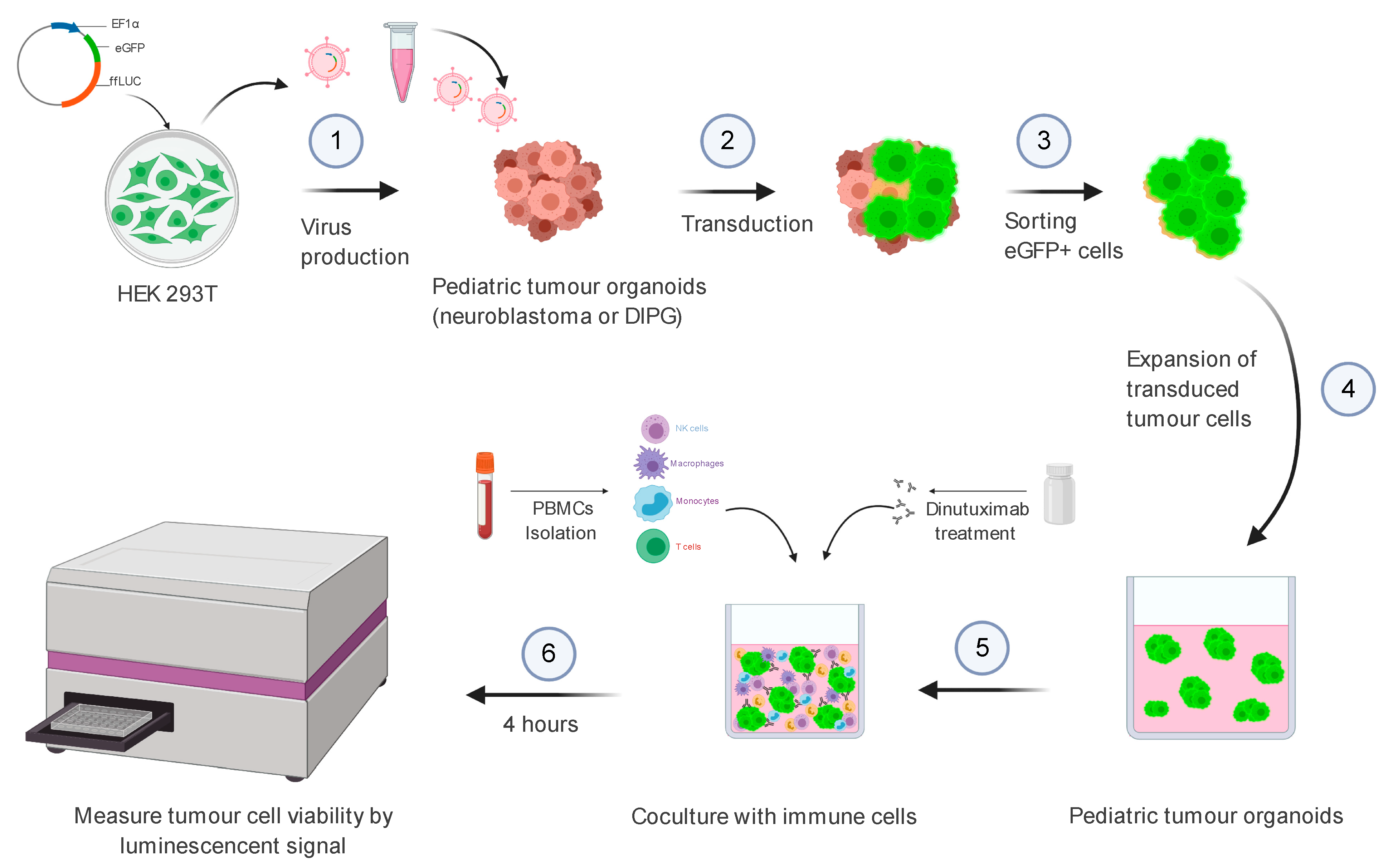
2.6. Methods
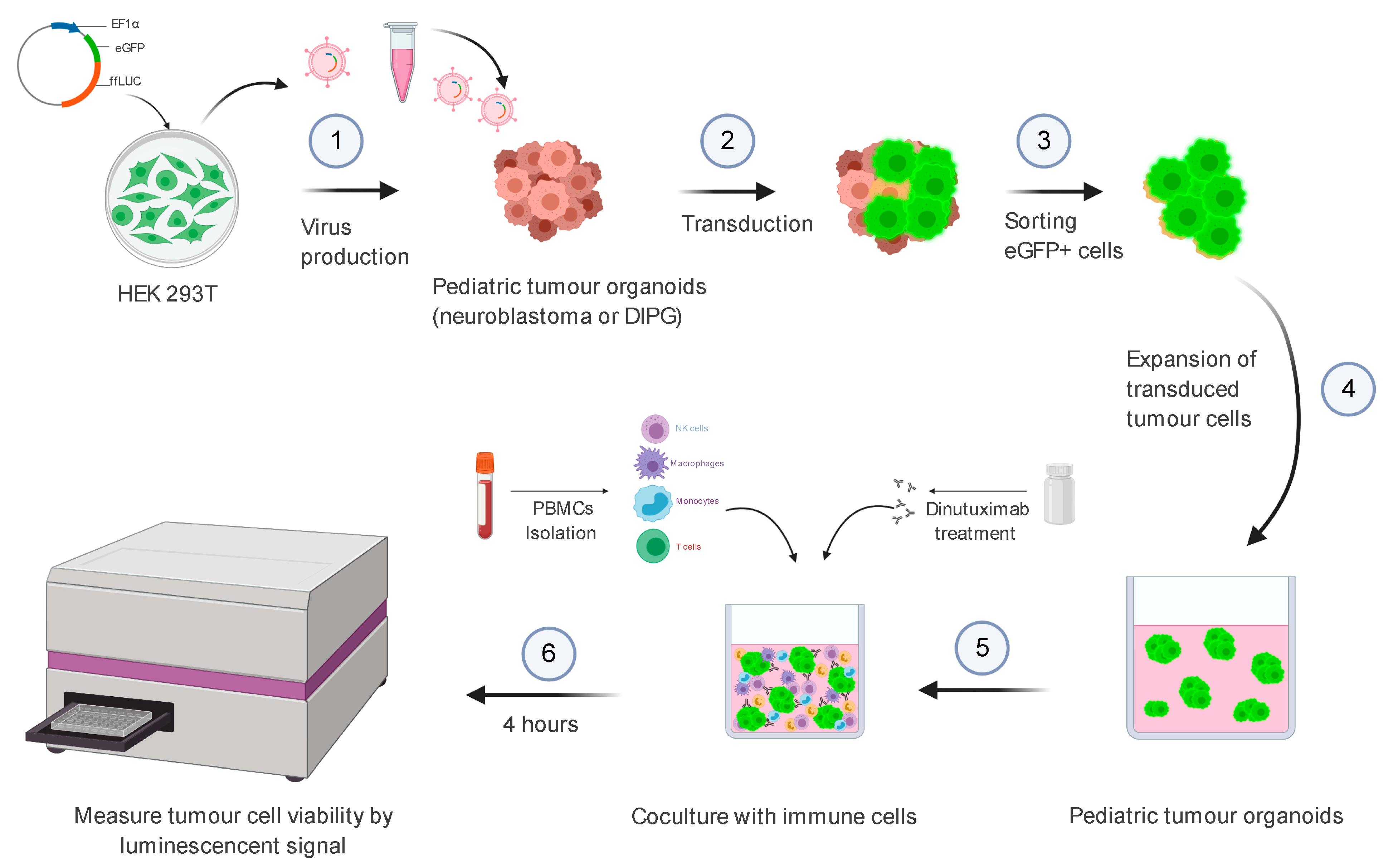
2.6.1. Part 1: Virus Production
Virus Production—Day 0
- Seed HEK293T cells in a 145/20 mm cell culture dish in 20 mL HEK293 medium to achieve 60–80% confluency the day after and place them in a CO2 incubator (5% CO2, 37 °C).
Virus Production—Day 1
- Four hours prior to transfection, refresh the HEK293T medium without detaching the adherent cells by removing 20 mL medium via the sides of the dish and adding 15 mL fresh medium.
- Transfer 315 μL of PEI (1 mg/mL) to a 15-mL conical tube containing 2 mL Opti-MEM and mix well by brief vortexing prior to 5 min incubation at RT.
- Transfer plasmid DNA (7.2 μg pHDMG (ENV), 3.6 μg pRC/CMV-rev1b, 3.6 μg pHDM Tat1b, 3.6 μg pHDM Hgpm2 and 45 μg of the transfer plasmid) to a 15 mL conical tube containing 3 mL Opti-MEM and mix well by brief vortexing.
- Add PEI dropwise to plasmid DNA and mix well by brief vortexing prior to 30 min incubation at RT.
- Transfer the PEI/DNA mixture dropwise to the cells and gently rotate the plate to equally distribute the PEI/plasmid DNA mixture in the plate and incubate the plate overnight in a CO2 incubator (5% CO2, 37 °C).
Virus Production—Day 2
- 6.
- Replace HEK293T medium with 20 mL fresh neuroblastoma organoid medium without detaching the adherent cells.
Virus Production—Day 3
- 7.
- Transfer all medium from the plate into a 50 mL conical tube for storage at 4 °C until further use in step 10.
- 8.
- Add 20 mL fresh neuroblastoma organoid medium to the HEK293T cells and incubate the cells for another 24 h.
Virus Production—Day 4
- 9.
- Remove all medium from the plate and transfer it into the same collecting tube used to harvest the previous day.
- 10.
- Pass the harvested medium through a 0.45 mm filter to remove any cell debris. Note: use only low protein binding filters such as cellulose acetate or polyethersulfone (PES).
2.6.2. Part 2: Concentrating Lentiviral Supernatants
2.6.3. Part 3: Virus Transduction of Patient-Derived Cells
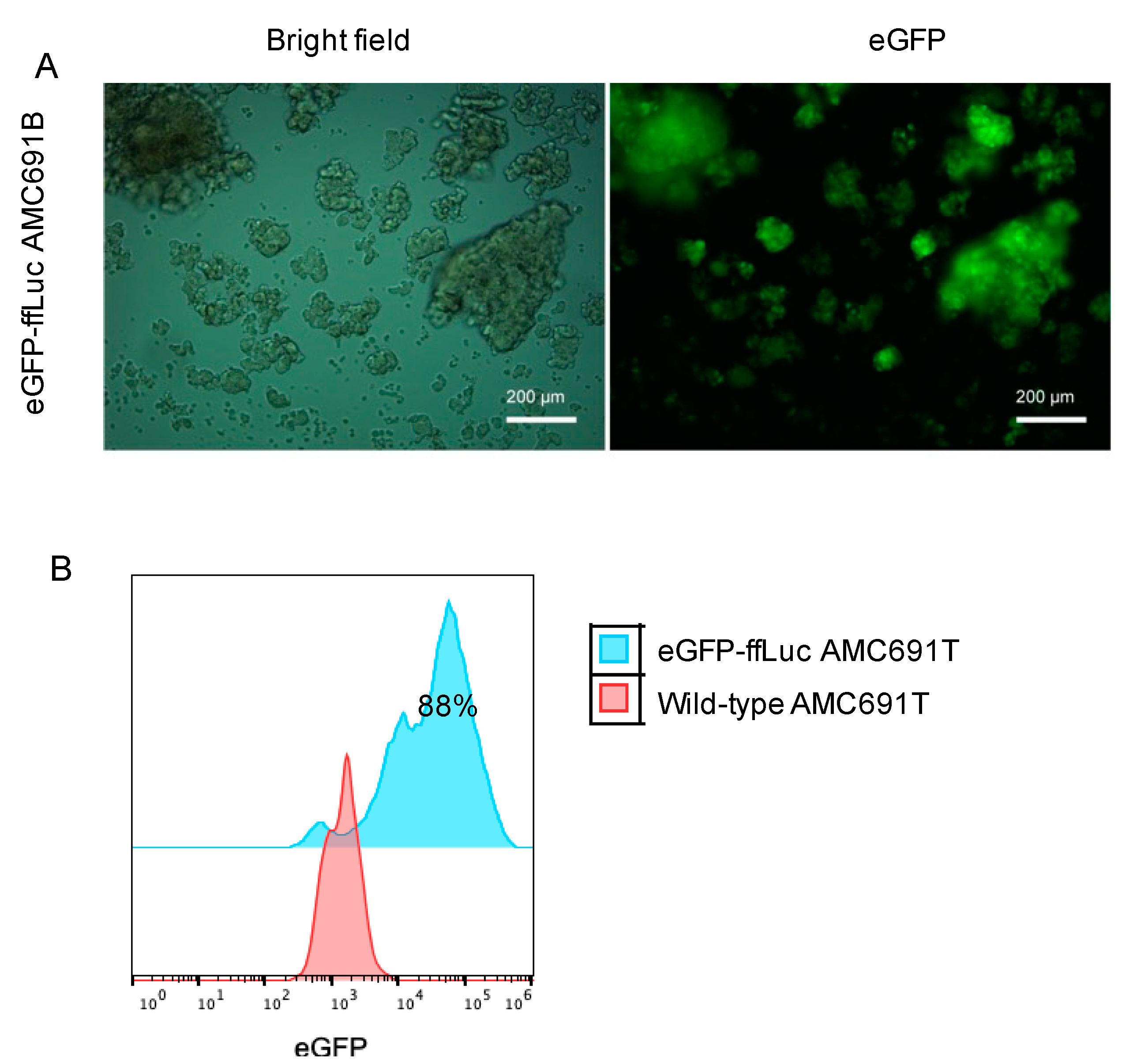
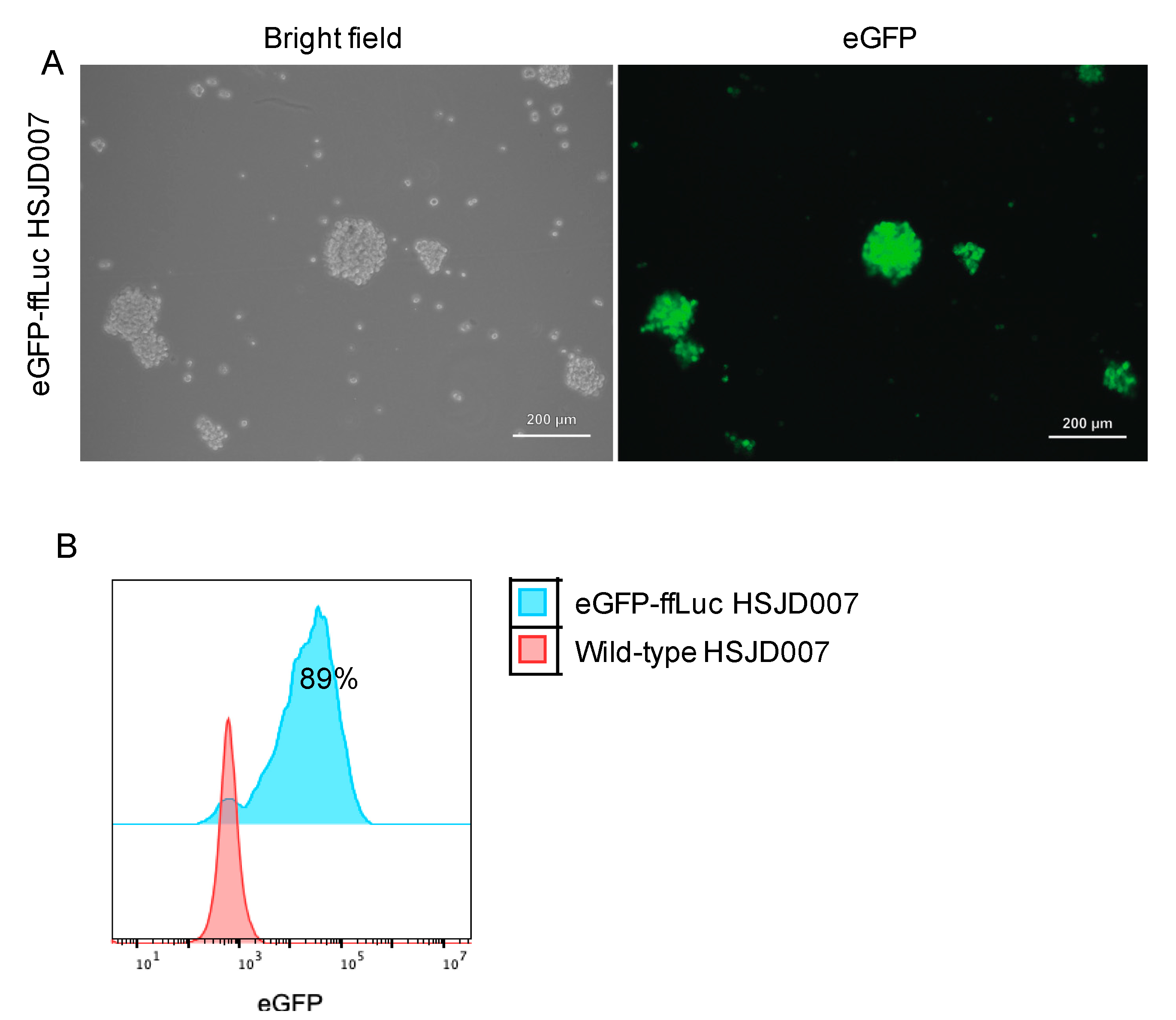
2.6.4. Part 4: Purification Transduced Cells by FACS
- 29.
- Transfer the transduced and matching wild type primary cells in separate 15 mL conical tubes.
- 30.
- Dissociate the cells with 1 mL Accutase until single-cell suspensions are obtained (check under the microscope).
- 31.
- Add 10 mL of serum-free culture medium to halt the dissociation reaction.
- 32.
- Centrifuge the tubes at 300 RCF for 5 min at 4 °C.
- 33.
- Aspirate and discard the supernatants.
- 34.
- Add 1 mL of serum-free culture medium to the tubes.
- 35.
- Transfer the cells into Falcon® Round-Bottom Tubes with Cell Strainer Cap.
- 36.
- Keep all tubes on ice and covered with aluminium foil to protect from light.
- 37.
- All samples are analysed and sorted on a Sony SH800 Cell Sorter.
2.6.5. Part 5: Antibody-Dependent Cellular Cytotoxicity (ADCC) Assay
Organoid Seeding—Day 0
- 38.
- Harvest the organoids from cell culture and transfer the organoid suspensions into a 50 mL conical tube.
- 39.
- Spin the tube at 300× g for 5 min.
- 40.
- Discard the supernatant medium and resuspend the organoid pellet in 1 mL Accutase.
- 41.
- Dissociate the organoids into single cells by pipetting up and down against the bottom of the 50 mL tubes.
- 42.
- Add 9 mL organoid medium to inactivate the Accutase and mix the solution by pipetting up and down.
- 43.
- Spin the tube at 300× g for 5 min.
- 44.
- Remove the supernatant medium using a suction pipette.
- 45.
- Resuspend the cell pellet in 1 mL organoid medium
- 46.
- Filter the single-cell organoid suspension by transferring the sample to FACS tubes.
- 47.
- Count the cells by mixing 10 µL of the single-cell suspension with 10 µL Trypan blue. Transfer 10 µL of the mixture into the counting slides on both sides A and B and count the cells.
- 48.
- Calculate the required volume to seed the optimal number of single cells (in this study, 40,000 cells/well) in 150 µL medium/well into a white 96-well plate.
- 49.
- Add 200 µL of organoid medium into the designated wells containing medium only.
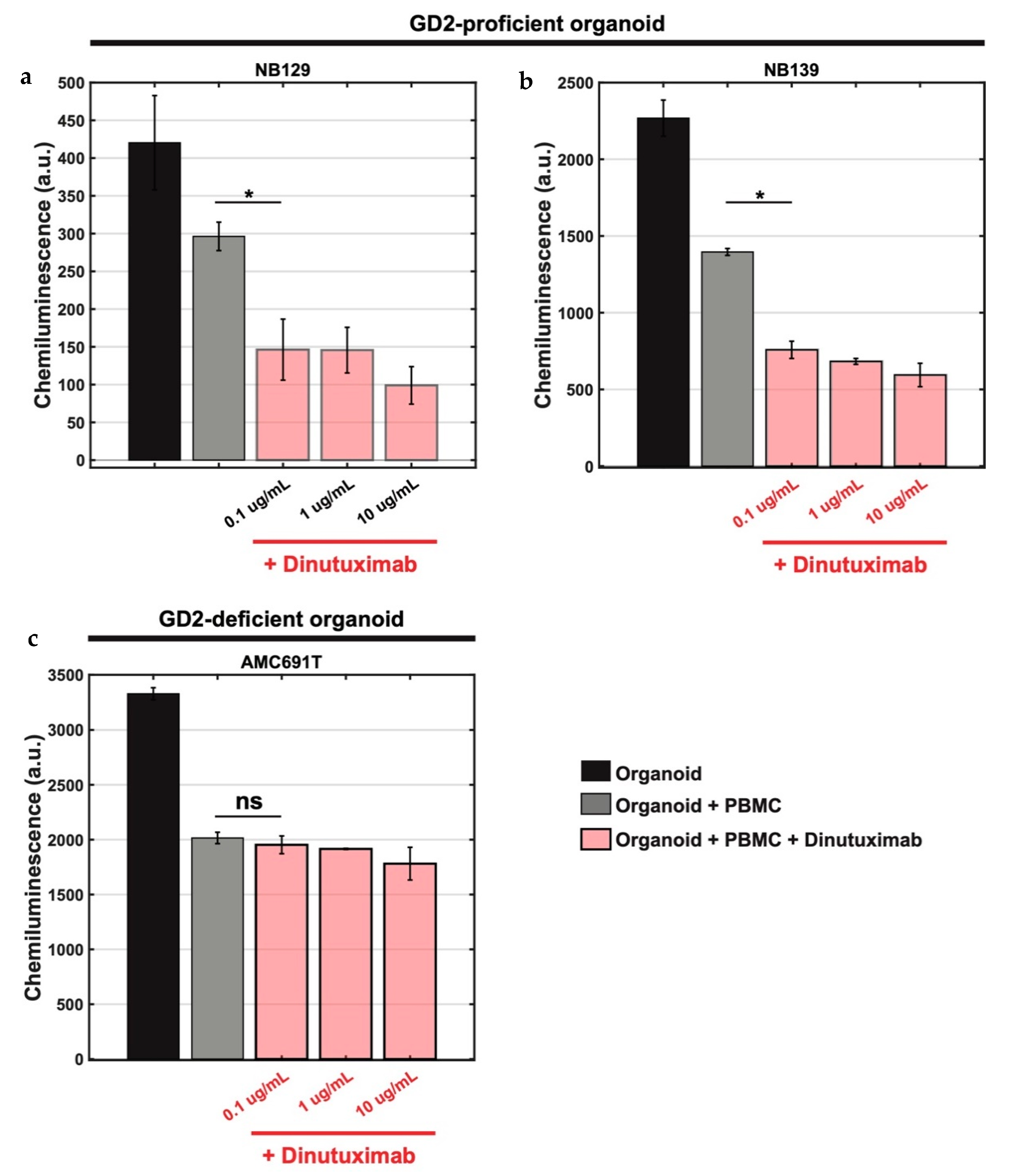
Dinutuximab Treatment Neuroblastoma Organoids—Day 0
- 50.
- For cytotoxicity studies, NB organoid lines were not treated in the absence or presence of PBMCs (controls) or treated with dinutuximab in the presence of PBMCs.
- 51.
- Add different concentrations of dinutuximab (10 µg/mL, 1 µg/mL, 100 ng/mL) to the neuroblastoma cells.
- 52.
- Place the 96-well plate into the incubator until the isolated PBMC cells are added.
Peripheral Blood Mononuclear Cell (PBMC) Isolation from Donor Blood—Day 0
- 53.
- PBMCs were isolated from the blood of healthy donors using the Lymphoprep Kit (Stemcell Technologies) according to the manufacturer’s protocol.
- 54.
- Isolated PBMCs washed with an equal amount of PBS + 2% FBS and resuspend the PBMC pellet in 2 mL organoid medium.
- 55.
- Count the PBMCs and calculate the number of PBMCs needed to obtain the desired effector to target (E:T) ratio (here we used 20:1 E:T ratio, 800.000 PBMCs in 50 μL organoid medium per well).
Coculture of Organoids and PBMC—Day 0
- 56.
- Add 800,000 PBMCs in 50 µL organoid medium to each requested well.
- 57.
- Incubate the plate for 4 h in the incubator.
- 58.
- Add D-luciferin (1:200 dilution) to the wells.
- 59.
- Spin the plate at 300× g for 5 min.
- 60.
- Incubate the plate for 5 min in the incubator.
- 61.
- Wrap the plate in aluminium foil to protect from light and read out the bioluminescence signal on the plate reader (FLUOstar Omega).
3. Results
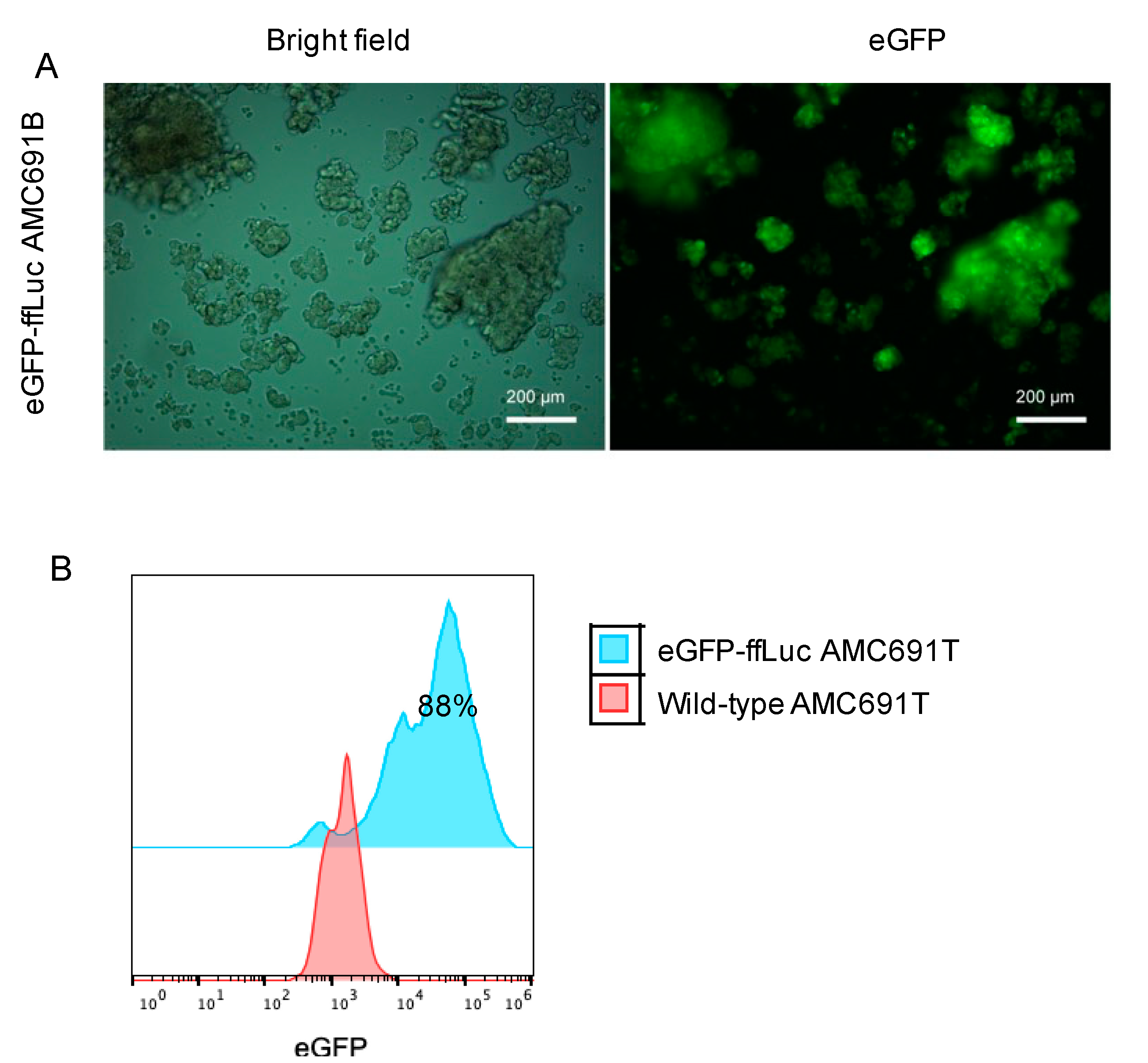
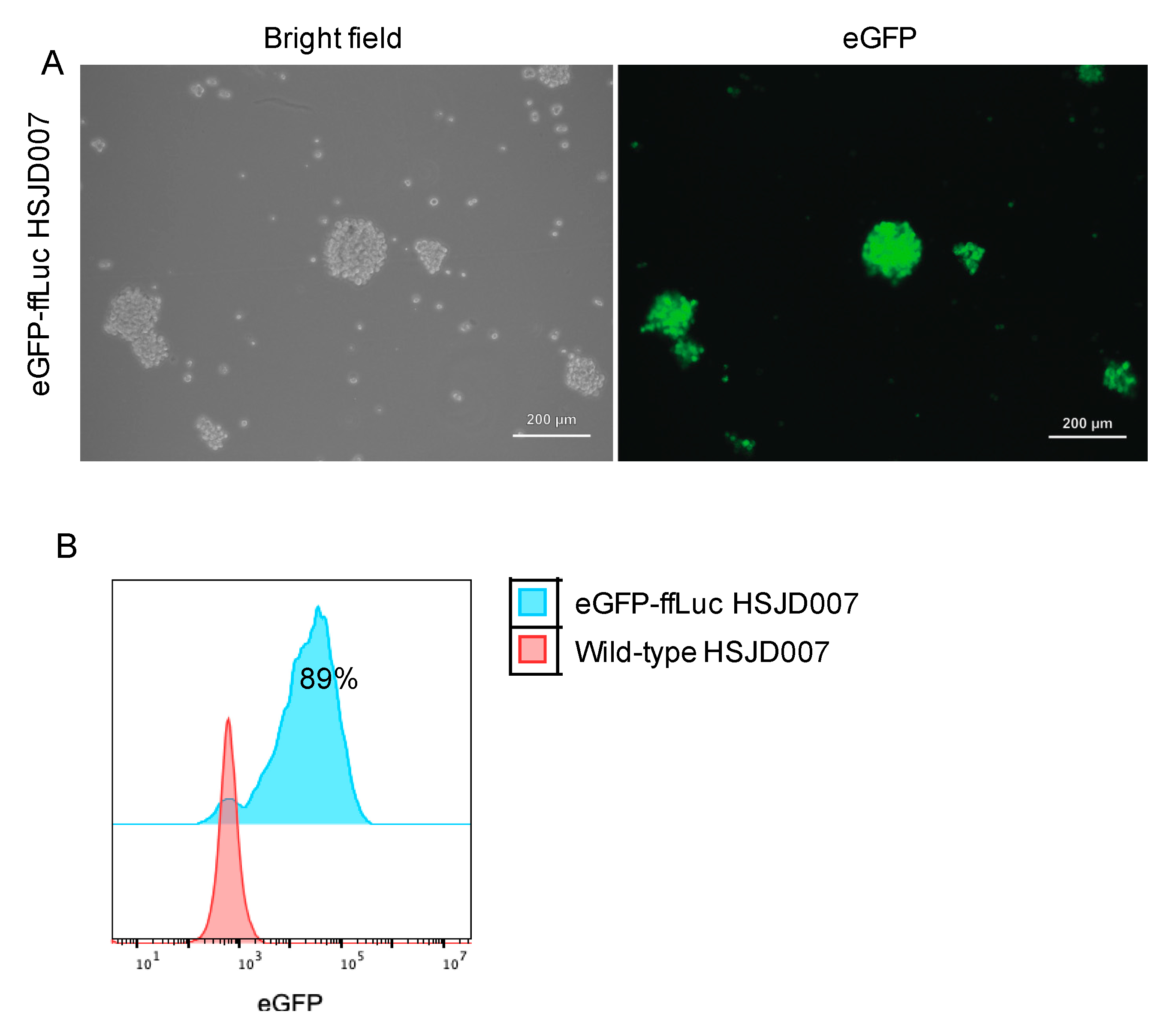
3.1. Transduction Efficiency of DIPG Neurospheres and Neuroblastoma Organoids
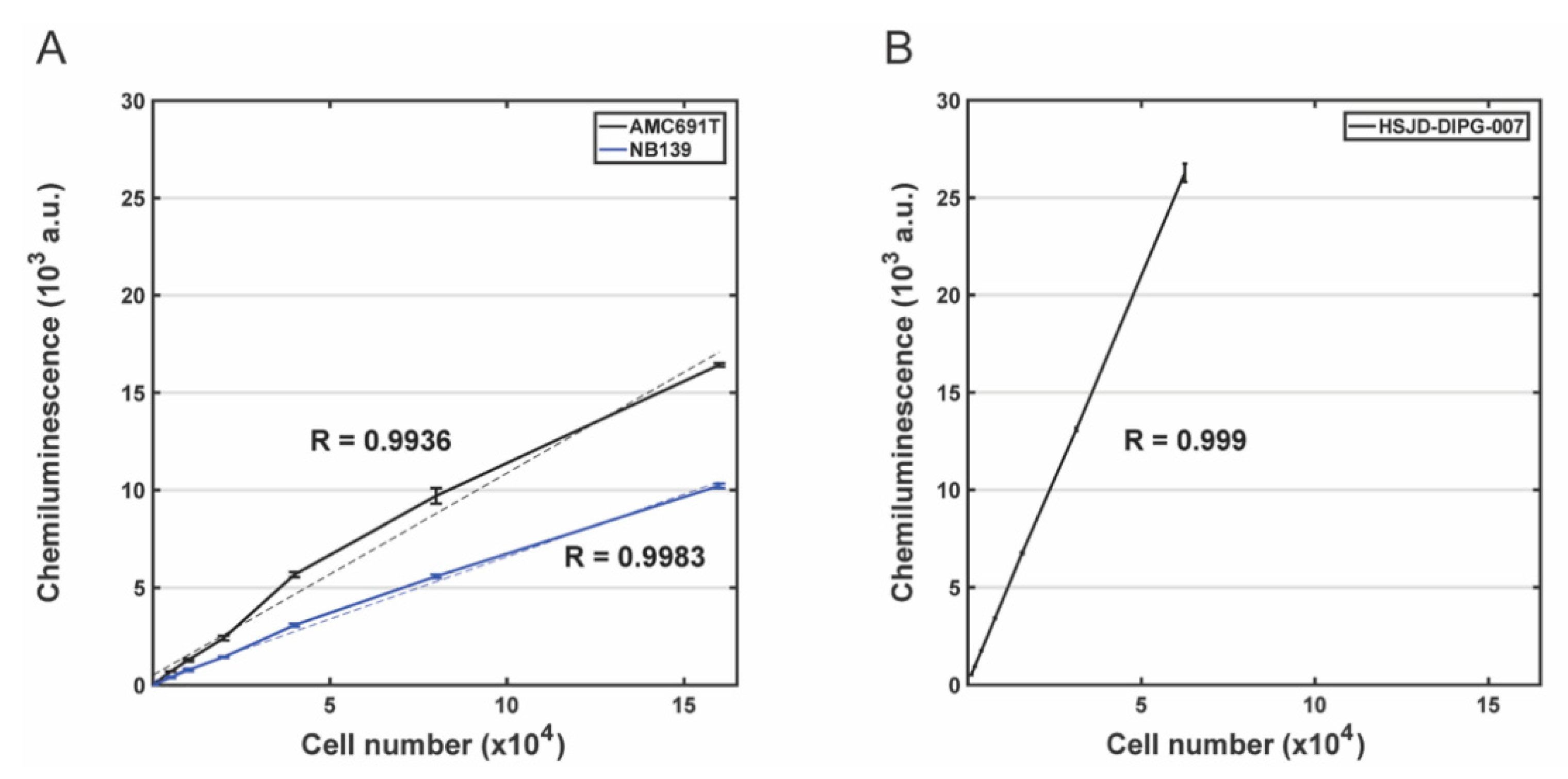
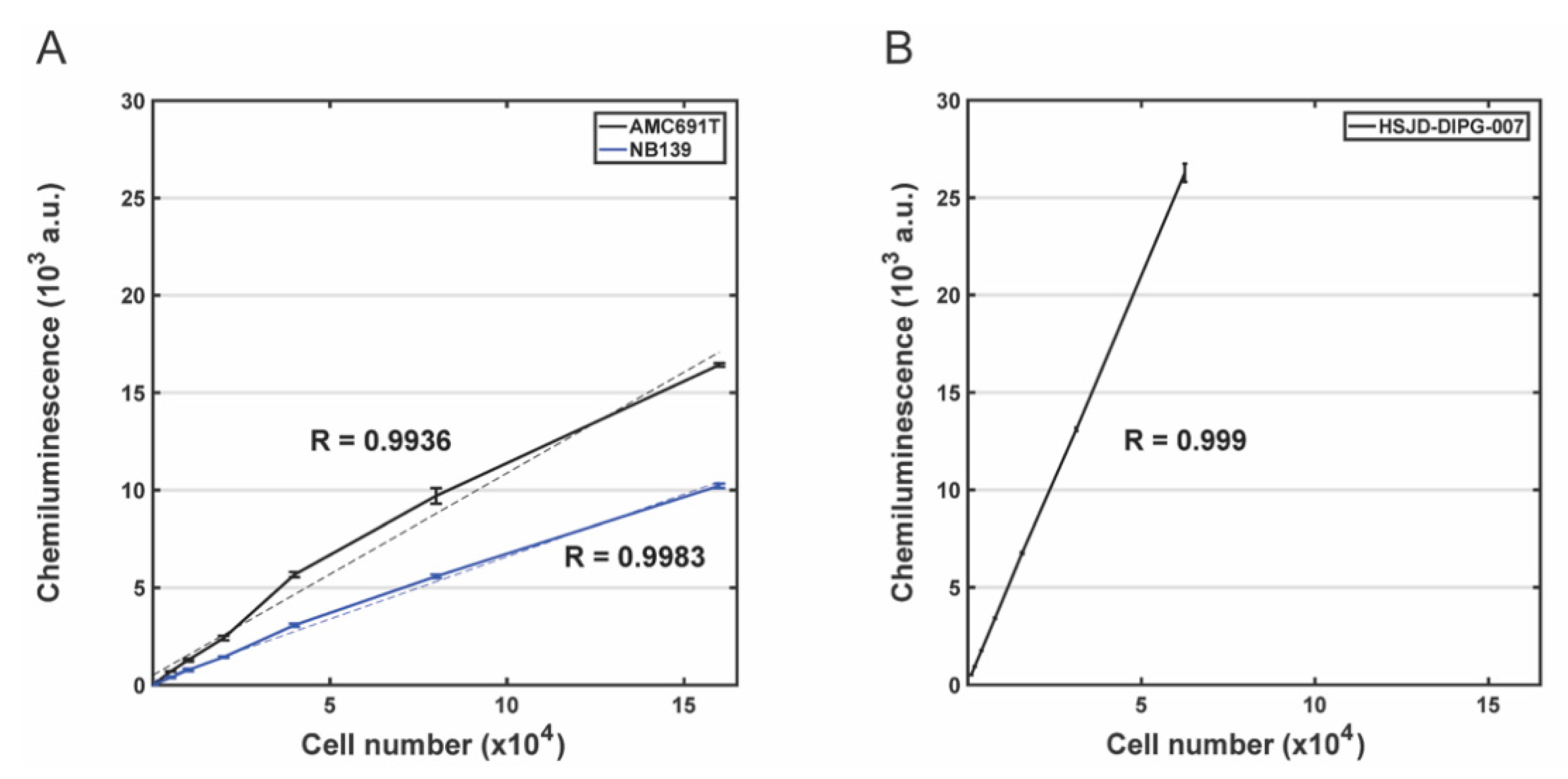
3.2. Evaluation of the Correlation between Bioluminescence Signal and the Number of Living Cells for DIPG Neurospheres and Neuroblastoma Organoids
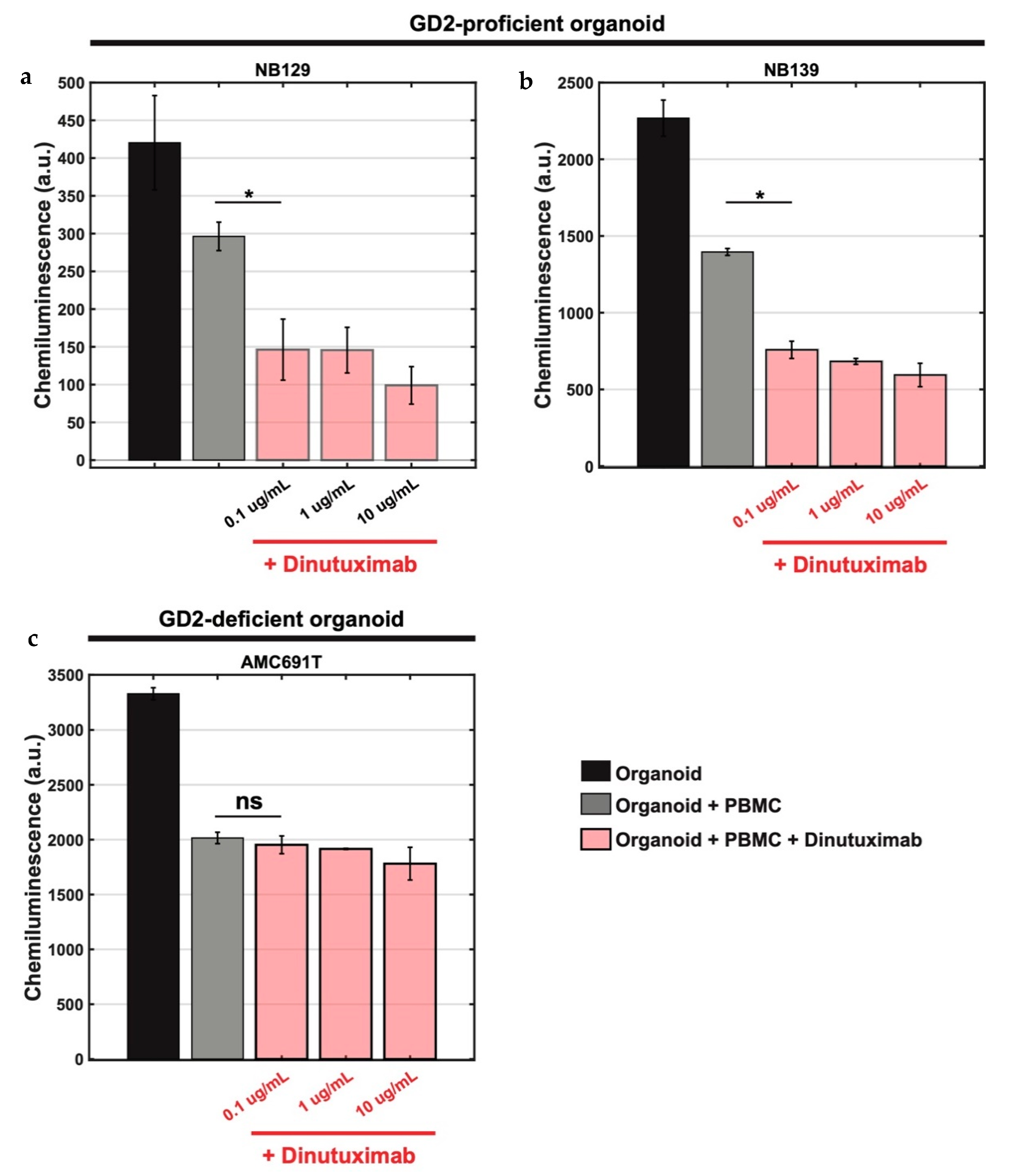
3.3. Bioluminescence-Based Ex Vivo Test Coculture System of Pediatric Tumour Organoids and PBMCs for Assessment of Anti-GD2 Immunotherapy
4. Discussion
5. Comparison with Other Methods
6. Biosafety Considerations
7. Technical Limitations
8. Applications of the Protocol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Marayati, R.; Quinn, C.H.; Beierle, E.A. Immunotherapy in Pediatric Solid Tumors—A Systematic Review. Cancers 2019, 11, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calandrini, C.; Schutgens, F.; Oka, R.; Margaritis, T.; Candelli, T.; Mathijsen, L.; Ammerlaan, C.; van Ineveld, R.L.; Derakhshan, S.; de Haan, S.; et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Fusco, P.; Parisatto, B.; Rampazzo, E.; Persano, L.; Frasson, C.; Di Meglio, A.; Leslz, A.; Santoro, L.; Cafferata, B.; Zin, A.; et al. Patient-derived organoids (PDOs) as a novel in vitro model for neuroblastoma tumours. BMC Cancer 2019, 19, 970. [Google Scholar] [CrossRef]
- Xu, C.; Liu, X.; Geng, Y.; Bai, Q.; Pan, C.; Sun, Y.; Chen, X.; Yu, H.; Wu, Y.; Zhang, P.; et al. Patient-derived DIPG cells preserve stem-like characteristics and generate orthotopic tumours. Oncotarget 2017, 8, 76644. [Google Scholar] [CrossRef] [Green Version]
- Lohmueller, J.; Finn, O.J. Current modalities in cancer immunotherapy: Immunomodulatory antibodies, CARs and vaccines. Pharmacol. Ther. 2017, 178, 31–47. [Google Scholar] [CrossRef]
- Wonderlich, J.; Shearer, G.; Livingstone, A.; Brooks, A.; Soloski, M.J.; Presby, M.M. Induction and measurement of cytotoxic T lymphocyte activity. Curr. Protoc. Immunol. 2018, 120, 3–11. [Google Scholar] [CrossRef]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity assays: In vitro methods to measure dead cells. In Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Indianapolis, IN, USA, 2019. [Google Scholar]
- Węsierska-Gądek, J.; Gueorguieva, M.; Ranftler, C.; Zerza-Schnitzhofer, G. A new multiplex assay allowing simultaneous detection of the inhibition of cell proliferation and induction of cell death. J. Cell. Biochem. 2005, 96, 1–7. [Google Scholar] [CrossRef]
- Goers, L.; Freemont, P.; Polizzi, K.M. Coculture systems and technologies: Taking synthetic biology to the next level. J. R. Soc. Interface 2014, 11, 20140065. [Google Scholar] [CrossRef] [Green Version]
- Contag, C.H.; Bachmann, M.H. Advances in vivo bioluminescence imaging of gene expression. Annu. Rev. Biomed. Eng. 2002, 4, 235–260. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.A.; Lee, E.; Bachmann, M.H.; Salicioni, A.M.; Behrens, E.M.; Kambayashi, T.; Baldwin, C.L. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS ONE. 2014, 9, e89357. [Google Scholar] [CrossRef] [PubMed]
- Krinner, S.; Heitzer, A.; Asbach, B.; Wagner, R. Interplay of promoter usage and intragenic CpG content: Impact on GFP reporter gene expression. Hum. Gene Ther. 2015, 26, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Boult, J.K.; Taylor, K.R.; Vinci, M.; Popov, S.; Jury, A.; Molinari, V.; Alonso, M.M.; Ingram, W.; Caraboso, A.M.; Monje, M.; et al. Novel orthotopic pediatric high-grade glioma xenografts evaluated with magnetic resonance imaging mimic human disease. In Proceedings of the AACR 106th Annual Meeting 2015, Philadelphia, PA, USA, 18–22 April 2015. [Google Scholar]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Cockle, J.V.; Picton, S.; Levesley, J.; Ilett, E.; Carcaboso, A.M.; Short, S.; Steel, L.P.; Melcher, A.; Lawler, S.E.; Brüning-Richardson, A. Cell migration in paediatric glioma; characterization and potential therapeutic targeting. Br. J. Cancer 2015, 112, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef]
- Meel, M.H.; de Gooijer, M.C.; Metselaar, D.S.; Sewing, A.C.; Zwaan, K.; Waranecki, P.; Breur, M.; Buil, L.C.; Lagerweij, T.; Wedekind, L.E.; et al. The combined therapy of AXL and HDAC inhibition reverses mesenchymal transition in diffuse intrinsic pontine glioma. Clin. Cancer Res. 2020, 26, 3319–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 Inhibitors Display Beneficial Effects in Preclinical Models of Mutant Diffuse Intrinsic Pontine Glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef]
- Sewing, A.C.; Lagerweij, T.; van Vuurden, D.G.; Meel, M.H.; Veringa, S.J.; Carcaboso, A.M.; Gaillard, P.J.; Vandertop, W.P.; Wesseling, P.; Noske, D.; et al. Preclinical evaluation of convection-enhanced delivery of liposomal doxorubicin to treat pediatric diffuse intrinsic pontine glioma and thalamic high-grade glioma. J. Neurosurg. Pediatr. 2017, 19, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Preclinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [Green Version]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2019, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid models of tumor immunology. Trends Immunol. 2020, 41, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Meel, M.H.; Metselaar, D.S.; Waranecki, P.; Kaspers, G.J.; Hulleman, E. An efficient method for the transduction of primary pediatric glioma neurospheres. MethodsX 2018, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Yan, T.; Zhu, H.; Liang, X.; Leiss, L.; Sakariassen, P.Ø.; Skaftnesmo, K.O.; Huang, B.; Costea, D.E.; Enger, P.Ø.; et al. A coculture model with brain tumor-specific bioluminescence demonstrates astrocyte-induced drug resistance in glioblastoma. J. Transl. Med. 2014, 12, 1–9. [Google Scholar] [CrossRef]
- Hannah, R.; Beck, M.; Moravec, R.; Riss, T. CellTiter-Glo™ Luminescent cell viability assay: A sensitive and rapid method for determining cell viability. Promega Cell Notes 2001, 2, 11–13. [Google Scholar]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Liu, Y.; Colberg-Poley, A.M.; Kaucic, K.; Ladisch, S. Induction of GM1a/GD1b synthase triggers complex ganglioside expression and alters neuroblastoma cell behavior; a new tumor cell model of ganglioside function. Glycoconj. J. 2011, 28, 137. [Google Scholar] [CrossRef] [Green Version]
- Schulz, G.; Cheresh, D.A.; Varki, N.M.; Yu, A.; Staffileno, L.K.; Reisfeld, R.A. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. 1984, 44, 5914–5920. [Google Scholar]
- Cheung, N.K.; Saarinen, U.M.; Neely, J.E.; Landmeier, B.; Donovan, D.; Coccia, P.F. Monoclonal antibodies to a glycolipid antigen on human neuroblastoma cells. Cancer Res. 1985, 45, 2642–2649. [Google Scholar]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Transgene | Vector | Description |
|---|---|---|
| FfLuc, eGFP | pJ01668 | epHIV7 vector, firefly luciferase, T2A sequence, eGFP |
| pHDM-Hgpm2 | Packaging plasmid for lentiviral vector | |
| pRC/CMV-rev1b | Regulatory plasmid for lentiviral vector | |
| pHDMG | Envelope plasmid used for lentivirus production | |
| pHDM-tat1b | Regulatory plasmid for lentiviral vector |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Kholosy, W.; Derieppe, M.; van den Ham, F.; Ober, K.; Su, Y.; Custers, L.; Schild, L.; M. J. van Zogchel, L.; M. Wellens, L.; R. Ariese, H.; et al. Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies. J. Pers. Med. 2021, 11, 869. https://doi.org/10.3390/jpm11090869
M. Kholosy W, Derieppe M, van den Ham F, Ober K, Su Y, Custers L, Schild L, M. J. van Zogchel L, M. Wellens L, R. Ariese H, et al. Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies. Journal of Personalized Medicine. 2021; 11(9):869. https://doi.org/10.3390/jpm11090869
Chicago/Turabian StyleM. Kholosy, Waleed, Marc Derieppe, Femke van den Ham, Kim Ober, Yan Su, Lars Custers, Linda Schild, Lieke M. J. van Zogchel, Lianne M. Wellens, Hendrikus R. Ariese, and et al. 2021. "Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies" Journal of Personalized Medicine 11, no. 9: 869. https://doi.org/10.3390/jpm11090869
APA StyleM. Kholosy, W., Derieppe, M., van den Ham, F., Ober, K., Su, Y., Custers, L., Schild, L., M. J. van Zogchel, L., M. Wellens, L., R. Ariese, H., Szanto, C. L., Wienke, J., Dierselhuis, M. P., van Vuurden, D., Dolman, E. M., & Molenaar, J. J. (2021). Neuroblastoma and DIPG Organoid Coculture System for Personalized Assessment of Novel Anticancer Immunotherapies. Journal of Personalized Medicine, 11(9), 869. https://doi.org/10.3390/jpm11090869