
Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity


 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Primary AML Cells
2.2. Normal Cells
2.3. AML Cell Culture Medium
2.4. Reagents
2.5. In Vitro Cell Culture Studies
2.6. RNA Preparation, Labeling, and Microarray Hybridization
2.7. Flow Cytometric Analyses of Cell Viability
2.8. Mutational Analyses
2.9. Proteomic Analyses of Primary Human AML Cells
2.10. Statistical and Bioinformatical Analyses
3. Results
3.1. The Patient Population
3.2. Initial In Vitro Screening of the Antiproliferative Effects of Chloroquine on AML Primary Cells and Mononuclear Umbilical Cord-Derived Cells
3.3. AML Cell Proliferation Is Inhibited by Chloroquine Alone and in Combination with Cytarabine
3.4. Proteomic Comparison of Primary AML Cells Derived from Patients with High and Low Susceptibility to Chloroquine
3.5. Chloroquine Inhibits AML Cell Proliferation in Cocultures with MSCs
3.6. An Antiproliferative Effect of Chloroquine Is Detected for Most Patients and Even for Patients Insensitive to Cytarabine
3.7. The Antiproliferative Effect of Chloroquine on AML Cells Is Associated with a Distinct Gene Expression Profile
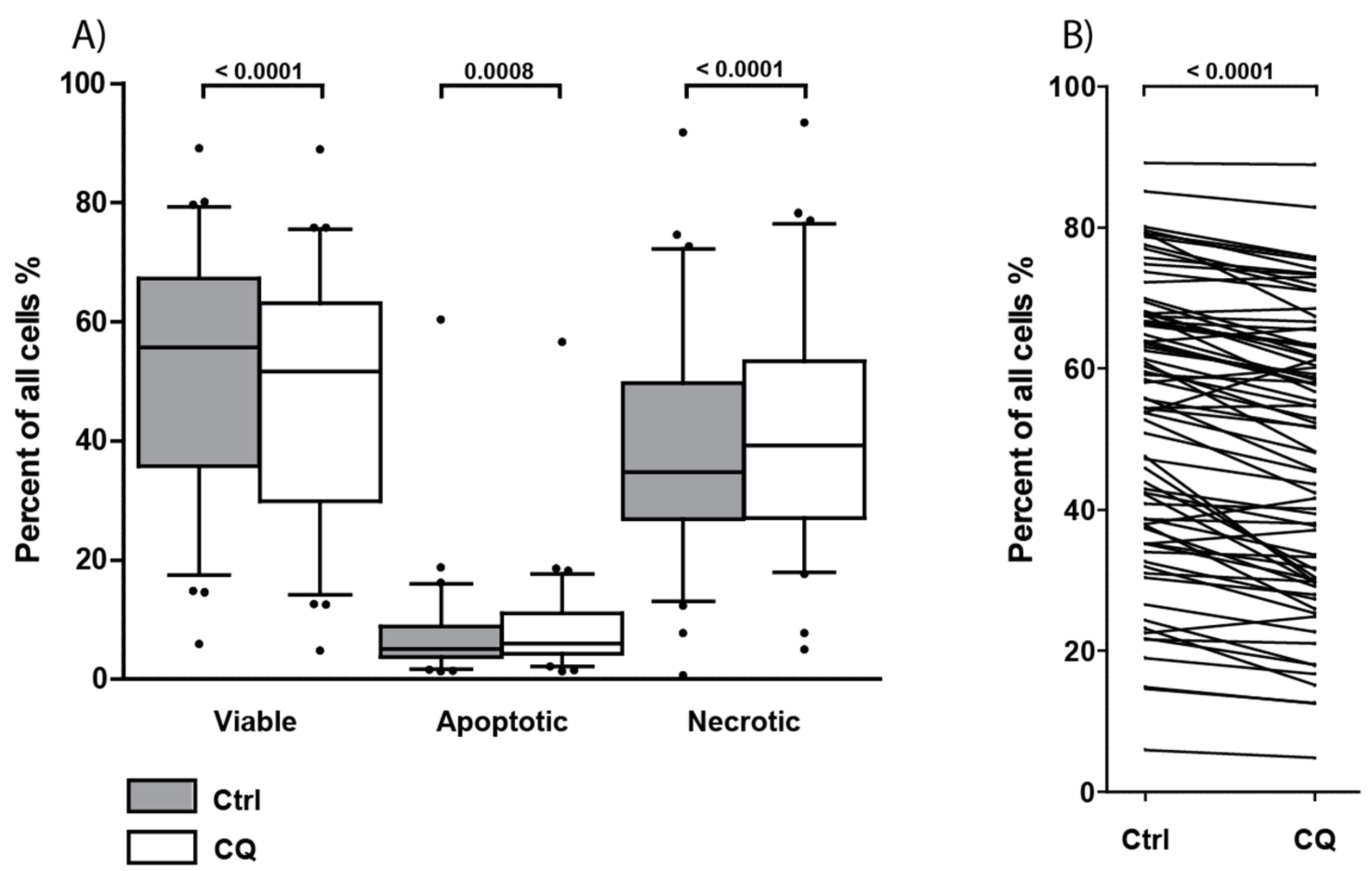
3.8. Treatment with Chloroquine Significantly Decreased Primary AML Cell Viability and Increased Apoptosis and Necrosis

3.9. Chloroquine Alters the Constitutive AML Cell Release of Only a Few Soluble Mediators by Primary Human AML Cells

3.10. High Chloroquine-Mediated Soluble Mediator Release Shows a Gene Expression Profile Associated with Genes Involved in Metabolic Processes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estey, E.H. Acute myeloid leukemia: 2021 update on risk-stratification and management. Am. J. Hematol. 2020, 95, 1368–1398. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Ramos, F. Acute myeloid leukemia in the older adults. Leuk. Res. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta 2015, 1853, 14–26. [Google Scholar] [CrossRef]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef]
- Du, W.; Xu, A.; Huang, Y.; Cao, J.; Zhu, H.; Yang, B.; Shao, X.; He, Q.; Ying, M. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [PubMed]
- Visser, N.; Lourens, H.J.; Huls, G.; Bremer, E.; Wiersma, V.R. Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine. Int. J. Mol. Sci. 2021, 22, 2337. [Google Scholar] [CrossRef]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting pharmacological effects of chloroquine in cancer treatment: Interference with inflammatory signaling pathways. Immunology 2020, 159, 257–278. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Gjertsen, B.T.; von Volkman, H.L. In vitro culture of human acute myelogenous leukemia (AML) cells in serum-free media: Studies of native AML blasts and AML cell lines. J. Hematother. Stem Cell Res. 2000, 9, 923–932. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Hovland, R.; Wergeland, L.; Huang, T.S.; Gjertsen, B.T. Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of Flt3-ligand effects in AML cell populations with and without genetic Flt3 abnormalities. Haematologica 2003, 88, 416–428. [Google Scholar] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar]
- Stapnes, C.; Doskeland, A.P.; Hatfield, K.; Ersvaer, E.; Ryningen, A.; Lorens, J.B.; Gjertsen, B.T.; Bruserud, Ø. The proteasome inhibitors bortezomib and PR-171 have antiproliferative and proapoptotic effects on primary human acute myeloid leukaemia cells. Br. J. Haematol. 2007, 136, 814–828. [Google Scholar] [CrossRef]
- Brenner, A.K.; Nepstad, I.; Bruserud, Ø. Mesenchymal Stem Cells Support Survival and Proliferation of Primary Human Acute Myeloid Leukemia Cells through Heterogeneous Molecular Mechanisms. Front. Immunol. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.; Ryningen, A.; Corbascio, M.; Bruserud, Ø. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int. J. Cancer 2006, 119, 2313–2321. [Google Scholar] [CrossRef]
- Stavrum, A.K.; Petersen, K.; Jonassen, I.; Dysvik, B. Analysis of Gene-Expression Data Using J-Express. Curr. Prot. Bioinform. 2008, 21, 7.3.1–7.3.25. [Google Scholar] [CrossRef]
- Egolf, S.; Aubert, Y.; Doepner, M.; Anderson, A.; Maldonado-Lopez, A.; Pacella, G.; Lee, J.; Ko, E.K.; Zou, J.; Lan, Y.; et al. LSD1 Inhibition Promotes Epithelial Differentiation through Derepession of Fate-Determining Transcription Factors. Cell Rep. 2019, 28, 1981–1992. [Google Scholar] [CrossRef]
- Ryningen, A.; Ersvaer, E.; Oyan, A.M.; Kalland, K.H.; Vintermyr, O.K.; Gjertsen, B.T.; Bruserud, Ø. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML) cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk. Res. 2006, 30, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Hovland, R.; Forthun, R.B.; Erdal, S.; Gjertsen, B.T.; Fredly, H.; Bruserud, Ø. Disease-stabilizing treatment based on all-trans retinoic acid and valproic acid in acute myeloid leukemia—Identification of responders by gene expression profiling of pretreatment leukemic cells. BMC Cancer 2017, 17, 630. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Døskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hiljkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Rreggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef] [PubMed]
- Nosál, R.; Jancinová, V. Cationic amphiphilic drugs and platelet phospholipase A(2) (cPLA(2)). Thromb. Res. 2002, 105, 339–345. [Google Scholar] [CrossRef]
- Maes, H.; Kuchnio, A.; Carmeliet, P.; Agostinis, P. How to teach an old dog new tricks: Autophagy-independent action of chloroquine on the tumor vasculature. Autophagy 2014, 10, 2082–2084. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, N.; Zhong, L.; Liu, L.; Fang, Y.; Qi, X.; Cao, J.; Yang, B.; He, Q.; Ying, M. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem. Pharmaco. 2014, 89, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Eom, J.I.; Jeung, H.K.; Jang, J.E.; Kim, J.S.; Cheong, J.W.; Kim, Y.S.; Min, Y.H. Induction of cytosine arabinoside-resistant human myeloid leukemia cell death through autophagy regulation by hydroxychloroquine. Biomed. Pharm. 2015, 73, 87–96. [Google Scholar] [CrossRef]
- Kazemi, A.; Sadri, M.; Houshmand, M.; Yazdi, N.; Zarif, M.N.; Anjam-Najmedini, A.; Tavakoli, R.; Ojaghi, M.; Ajami, M.; Ajami, M.; et al. The anticancer effects of pharmacological inhibition of autophagy in acute erythroid leukemia cells. Anticancer Drugs 2018, 29, 944–955. [Google Scholar] [CrossRef]
- Cheong, J.W.; Kim, Y.; Eom, J.I.; Jeung, H.K.; Min, Y.H. Enhanced autophagy in cytarabine arabinoside-resistant U937 leukemia cells and its potential as a target for overcoming resistance. Mol. Med. Rep. 2016, 13, 3433–3440. [Google Scholar] [CrossRef]
- Yan, Z.W.; Hou, J.K.; He, W.; Fan, L.; Huang, Y. Chloroquine enhances cobalt chloride-induced leukemic cell differentiation via the suppression of autophagy at the late phase. Biochem. Biophys. Res. Commun. 2013, 430, 926–932. [Google Scholar] [CrossRef]
- Forsbeck, K.; Nilsson, K. Iron metabolism of established human hematopoietic cell lines in vitro. Exp. Cell Res. 1983, 144, 323–332. [Google Scholar] [CrossRef]
- Bridges, K.R.; Hoffman, K.E. The effects of ascorbic acid on the intracellular metabolism of iron and ferritin. J. Biol. Chem. 1986, 261, 14273–14277. [Google Scholar] [CrossRef]
- Krajewski, W.A. Alterations in the internucleosomal DNA helical twist in chromatin of human erythroleukemia cells in vivo influences the chromatin higher-order folding. FEBS Lett. 1995, 361, 149–152. [Google Scholar] [CrossRef]
- Chernof, D.; Taylor, K.S. Hydroxychloroquine-Induced Agranulocytosis. Arch. Dermatol. 1968, 97, 163–164. [Google Scholar] [CrossRef]
- Ette, E.I.; Essien, E.E.; Brown-Awala, E.E. Pharmacokinetics of chloroquine: Saliva and plasma levels relationship. Eur. J. Drug Metab. Pharm. 1986, 11, 275–281. [Google Scholar] [CrossRef]
- Augustijns, P.; Geusens, P.; Verbeke, N. Chloroquine levels in blood during chronic treatment of patients with rheumatoid arthritis. Eur. J. Clin. Pharmacol. 1992, 42, 429–433. [Google Scholar]
- Pharmacology of antimalarial drugs. In Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015; pp. 205–284.
- Hubeek, I.; Kaspers, G.-J.L.; Ossenkoppele, G.J.; Peters, G.J. Deoxynucleoside analogs in cancer therapy. In Cancer Drug Discovery and Development; Peter, G.J., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2006; pp. 119–152. [Google Scholar]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In Vitro culture of aml cells—the present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Oyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Doskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.H.; Hood, L.; et al. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002, 11, 469–481. [Google Scholar] [CrossRef]
- Aasebø, E.; Berven, F.S.; Hovland, R.; Døskeland, S.O.; Bruserud, Ø.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers 2020, 12, 1466. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Aasebø, E.; Hernandez-Valladares, M.; Tsykunova, G.; Reikvam, H. Therapeutic targeting of leukemic stem cells in acute myeloid leukemia—The biological background for possible strategies. Expert Opin. Drug Discov. 2017, 12, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Döhner, K.; Thiede, C.; Jahn, N.; Panina, E.; Gambietz, A.; Larson, R.A.; Prior, T.W.; Marcucci, G.; Jones, D.; Krauter, J.; et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 2020, 135, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Bruserud, Ø.; Selheim, F. The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives. Int. J. Mol. Sci. 2020, 21, 6830. [Google Scholar] [CrossRef]
- Mer, A.S.; Lindberg, J.; Nilsson, C.; Klevebring, D.; Wang, M.; Grönberg, H.; Lehmann, S.; Rantalainen, M. Expression levels of long non-coding RNAs are prognostic for AML outcome. J. Hematol. Oncol. 2018, 11, 52. [Google Scholar] [CrossRef]
- Stäubert, C.; Bhuiyan, H.; Lindahl, A.; Broom, O.J.; Zhu, Y.; Islam, S.; Linnarsson, S.; Lehtiö, J.; Nordström, A. Rewired metabolism in drug-resistant leukemia cells: A metabolic switch hallmarked by reduced dependence on exogenous glutamine. J. Biol. Chem. 2015, 290, 8348–8359. [Google Scholar] [CrossRef]
- Lazarevic, V.; Hörstedt, A.S.; Johansson, B.; Antunovic, P.; Billström, R.; Derolf, Å.; Lehmann, S.; Möllgård, L.; Peterson, S.; Stockelberg, D.; et al. Failure matters: Unsuccessful cytogenetics and unperformed cytogenetics are associated with a poor prognosis in a population-based series of acute myeloid leukaemia. Eur. J. Haematol. 2015, 94, 419–423. [Google Scholar] [CrossRef]
- Grønbæk, K.; Müller-Tidow, C.; Perini, G.; Lehmann, S.; Bach Treppendahl, M.; Mills, K.; Plass, C.; Schlegelberger, B.; European Genomics and Epigenomics Study on MDS and AML (EuGESMA), COST Action BM0801. A critical appraisal of tools available for monitoring epigenetic changes in clinical samples from patients with myeloid malignancies. Haematologica 2012, 97, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Reiter, A.; Schrappe, M.; Ludwig, W.D.; Lampert, F.; Harbott, J.; Henze, G.; Niemeyer, C.M.; Gadner, H.; Müller-Weihrich, S.; Ritter, J.; et al. Favorable outcome of B-cell acute lymphoblastic leukemia in childhood: A report of three consecutive studies of the BFM group. Blood 1992, 80, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Bartaula-Brevik, S.; Hernandez-Valladares, M.; Bruserud, Ø. Vacuolar ATPase as a possible therapeutic target in human acute myeloid leukemia. Expert Rev. Hematol. 2018, 11, 13–24. [Google Scholar] [CrossRef]
- Lesiak, A.; Narbutt, J.; Sysa-Jedrzejowska, A.; Lukamowicz, J.; McCauliffe, D.P.; Wózniacka, A. Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus 2010, 19, 683–688. [Google Scholar] [CrossRef]
- Park, J.; Choi, K.; Jeong, E.; Kwon, D.; Benveniste, E.N.; Choi, C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 2004, 47, 9–20. [Google Scholar] [CrossRef]
- Chang, T.C.; Hsu, M.F.; Wu, K.K. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 2015, 10, e0126537. [Google Scholar] [CrossRef]
- Geiger, H.; Van Zant, G. The aging of lympho-hematopoietic stem cells. Nat. Immunol. 2002, 3, 329–333. [Google Scholar] [CrossRef]
- Pelt, J.; Busatto, S.; Ferrari, M.; Thompson, E.A.; Mody, K.; Wolfram, J. Chloroquine and nanoparticle drug delivery: A promising combination. Pharmacol. Ther. 2018, 191, 43–49. [Google Scholar] [CrossRef]
- Njaria, P.M.; Okombo, J.; Njuguna, N.M.; Chibale, K. Chloroquine-containing compounds: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1003–1024. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Lee, H. Chloroquine-based hybrid molecules as promising novel chemotherapeutic agents. Eur. J. Pharmacol. 2015, 762, 472–486. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, P.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, D.; Feld, J.; Dougherty, M.; Czaplinska, T.; Sanchez, G.; Kremyanskaya, M.; Bar-Natan, M.; Shih, A.H.; Keyzner, A.; Mascarenhas, J. Venetoclax and hypomethylating agent combination therapy in acute myeloid leukemia secondary to a myeloproliferative neoplasm. Leuk. Res. 2020, 98, 106456. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: A single-centre, phase 2 trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Chua, C.C.; Roberts, A.W.; Reynolds, J.; Fong, C.Y.; Ting, S.B.; Salmon, J.M.; MacRaild, S.; Ivey, A.; Tiong, I.S.; Fleming, S.; et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined with Modified Intensive Chemotherapy. J. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Danielson, N.; Sengsayadeth, S.; Rasche, A.; Culos, K.; Gatwood, K.; Wyatt, H.; Chinratanalab, W.; Dholaria, B.; Ferrell, P.B.; et al. The use of venetoclax-based salvage therapy for post-hematopoietic cell transplantation relapse of acute myeloid leukemia. Am. J. Hematol. 2020, 95, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients with Acute Myeloid Leukemia: Results from a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Precision Medicine Treatment in Acute Myeloid Leukemia Is Not a Dream. Hemato 2021, 2, 131–154. [Google Scholar] [CrossRef]





{kind=link}
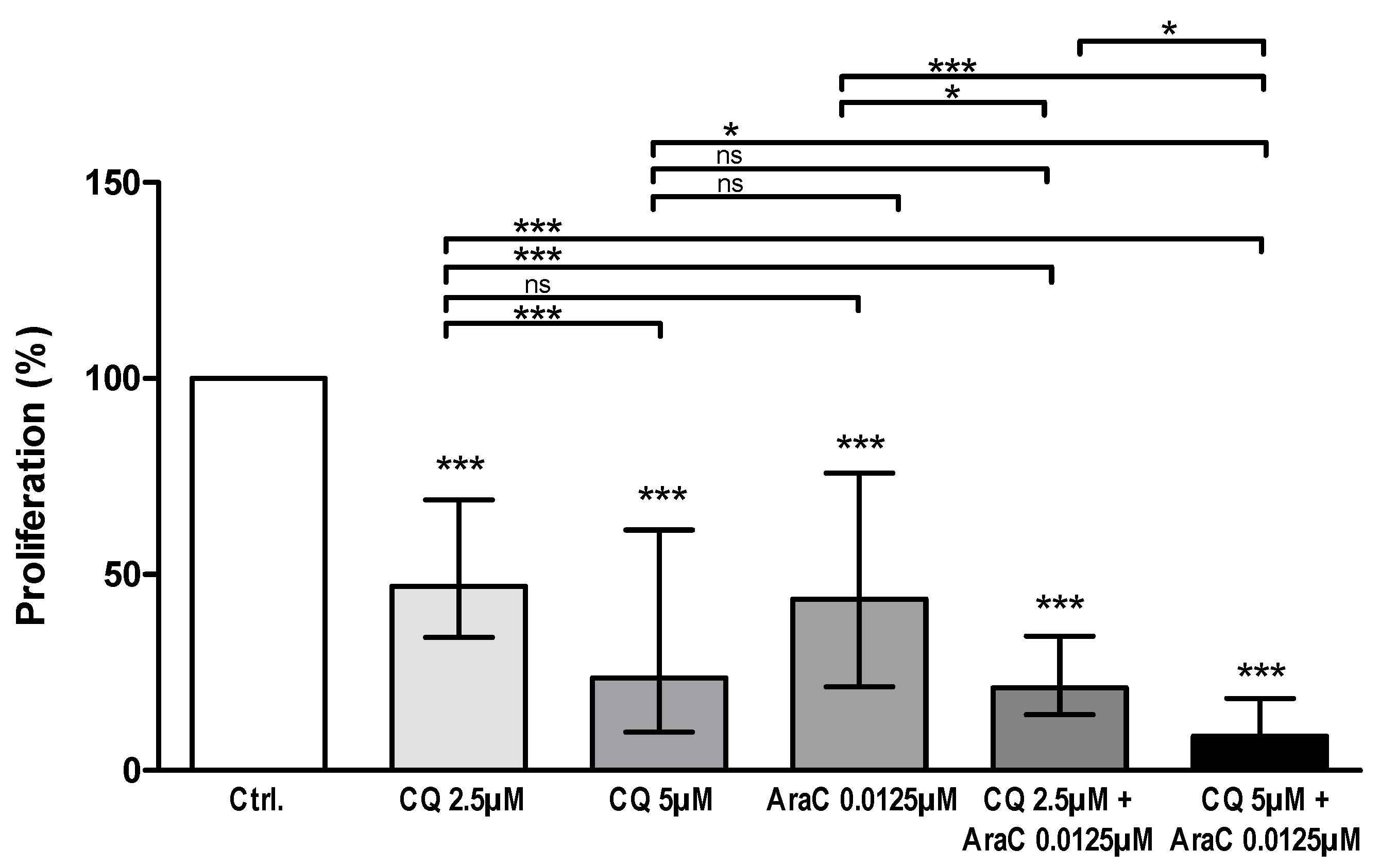{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Proteins (Referred to by Their Corresponding Gene Names) |
|---|---|
| Autophagy regulation | SIGIRR, PGPEP1, STK38L, DAP3, YBX1, CSDE1, PRMT1, HGSNAT, FAF1, FAM105A |
| Mitophagy regulation | ATPIF1, PGPEP1, STK38L |
| Cytoskeletal protein | DNAJC1, SIGIRR, TUBA1A, NUDCD3, TUBB6, TPPP3 |
| Intracellular trafficking | SYTL1, WDR81 |
| Endoplasmic reticulum | WDR81, LEPRE1, DPM3, CALU, SERPINH1, LY75 |
| Lysosomal protein | WDR81, LY75, HGSNAT |
| Mitochondria, metabolism | SARDH, SLC2A5, ATPIF1, H6PD, DAP3, MMS19, HK2 |
| Extracellular release | PPBP, YBX1 |
| Cell surface/adhesion | HLA-E, EPB41L2, ITGB3, ITGA2B, DPYSL3 |
| Intracellular signaling | DNAJC1, SIGIRR, STK38L, FAF1, TSTD1 |
| Transcription | SUGP2, SAP30L, PGPEP1, GTF2E2, NPM3, CRIP2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.S.; Tvedt, T.H.; Bruserud, Ø.; Hatfield, K.J. Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity. J. Pers. Med. 2021, 11, 779. https://doi.org/10.3390/jpm11080779
Grønningsæter IS, Reikvam H, Aasebø E, Bartaula-Brevik S, Hernandez-Valladares M, Selheim F, Berven FS, Tvedt TH, Bruserud Ø, Hatfield KJ. Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity. Journal of Personalized Medicine. 2021; 11(8):779. https://doi.org/10.3390/jpm11080779
Chicago/Turabian StyleGrønningsæter, Ida Sofie, Håkon Reikvam, Elise Aasebø, Sushma Bartaula-Brevik, Maria Hernandez-Valladares, Frode Selheim, Frode S. Berven, Tor Henrik Tvedt, Øystein Bruserud, and Kimberley Joanne Hatfield. 2021. "Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity" Journal of Personalized Medicine 11, no. 8: 779. https://doi.org/10.3390/jpm11080779
APA StyleGrønningsæter, I. S., Reikvam, H., Aasebø, E., Bartaula-Brevik, S., Hernandez-Valladares, M., Selheim, F., Berven, F. S., Tvedt, T. H., Bruserud, Ø., & Hatfield, K. J. (2021). Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity. Journal of Personalized Medicine, 11(8), 779. https://doi.org/10.3390/jpm11080779