
Sweat Chloride Testing and Nasal Potential Difference (NPD) Are Primary Outcome Parameters in Treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sweat Test
2.1. Physiological Principles and Relation to CFTR Activity
2.2. Analytical and Biological Variability
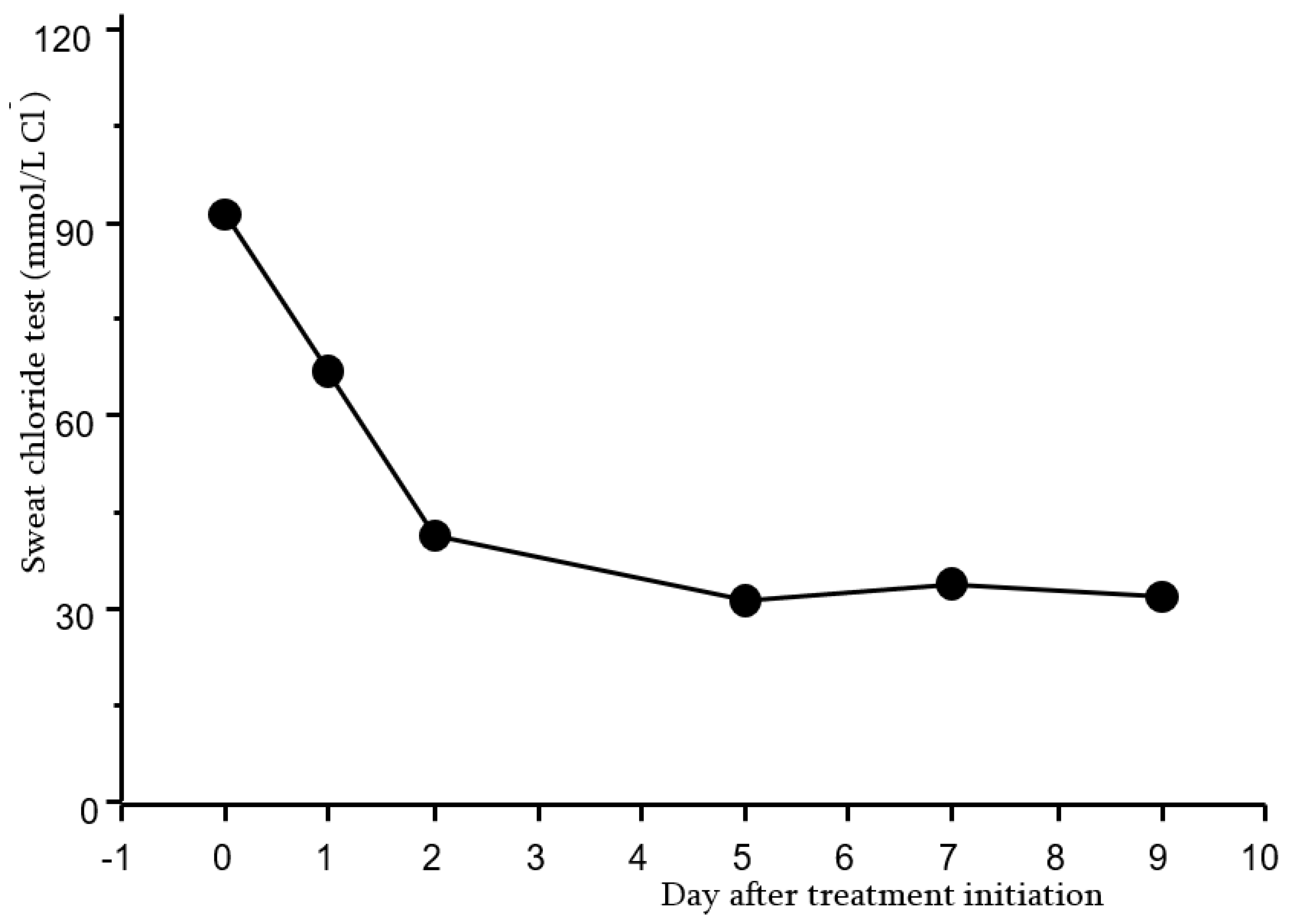
2.3. Sweat Test as a Primary Outcome in Clinical Trials with CFTR Modulator Therapies
3. Nasal Potential Difference
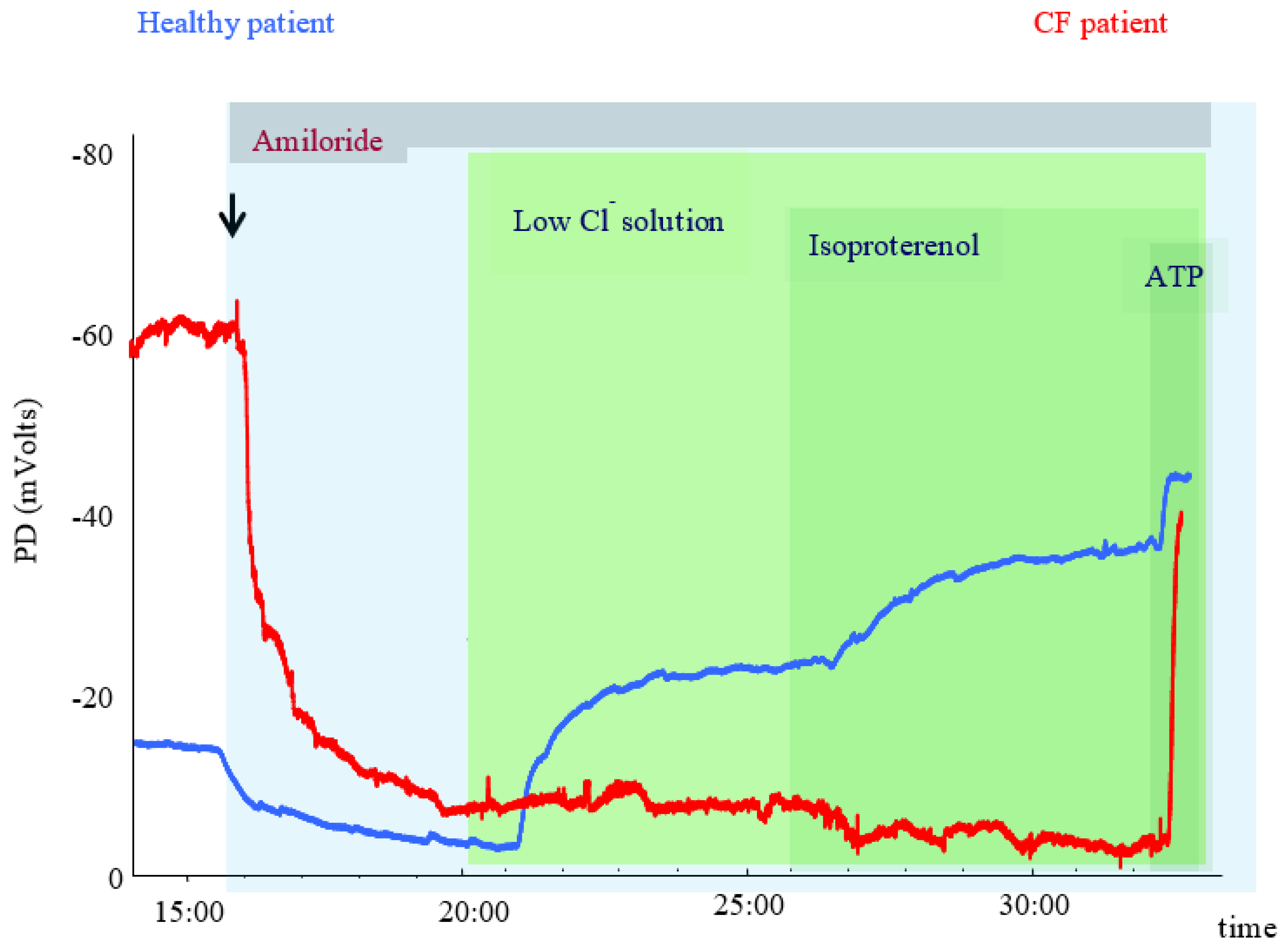
3.1. Physiological Principles and Relation to CFTR Activity
3.2. Analytical and Biological Variability
4. Clinical Trials
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Pranke, I.M.; Sermet-Gaudelus, I. Biosynthesis of cystic fibrosis transmembrane conductance regulator. Int. J. Biochem. Cell Biol. 2014, 52, 26–38. [Google Scholar] [CrossRef]
- Bareil, C.; Bergougnoux, A. CFTR gene variants, epidemiology and molecular pathology. Arch. Pediatr. 2020, 27 (Suppl. 1), eS8–eS12. [Google Scholar] [CrossRef]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Kent, L.; Davies, J.; Derichs, N.; Amaral, M.; Rowe, S.M.; Middleton, P.; de Jonge, H.; Bronsveld, I.; Wilschanski, M.; et al. European Cystic Fibrosis Society Clinical Trial Network Standardisation Committee. CFTR biomarkers: Time for promotion to surrogate endpoint. Eur. Respir. J. 2013, 41, 203–216. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Bel Stremler-Le, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef]
- Terlizzi, V.; Amato, F.; Castellani, C.; Ferrari, B.; Galietta, L.J.V.; Castaldo, G.; Taccetti, G. Ex vivo model predicted in vivo efficacy of CFTR modulator therapy in a child with rare genotype. Mol. Genet. Genom. Med. 2021, 9, e1656. [Google Scholar]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J., Jr.; Cystic Fibrosis Foundation. Diagnostic sweat testing: The Cystic Fibrosis Foundation guidelines. J. Pediatr. 2007, 151, 85–89. [Google Scholar] [CrossRef]
- Green, A.; Kirk, J.; on behalf of the Guidelines Development Group. Guidelines for the performance of the sweat test for the diagnosis of cystic fibrosis. Ann. Clin. Biochem. 2007, 44, 25–34. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef] [PubMed]
- Sweat Test: Stimulation and Collection European Cystic Fibrosis Society-Clinical Trial Network SOP 2018. Available online: ECFS-CTN@uzleuven.be (accessed on 8 May 2021).
- Cirilli, N.; Southern, K.W.; Buzzetti, R.; Barben, J.; Nährlich, L.; Munck, A.; Wilschanski, M.; De Boeck, K.; Derichs, N.; ECFS Diagnostic Network Working Group. Real life practice of sweat testing in Europe. J. Cyst. Fibros. 2017, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Van Goor, F.; Zha, J.; Stone, A.J.; Dong, Q.; Ordonez, C.L.; Rowe, S.M.; Clancy, J.P.; Konstan, M.W.; Hoch, H.E.; et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. 2014, 13, 139–147. [Google Scholar] [CrossRef]
- Munger, B.L.; Brusilow, S.W.; Cooke, R.E. An electron microscopic study of eccrine sweat glands in patients with cystic fibrosis of the pancreas. J. Pediatr. 1961, 59, 497–511. [Google Scholar] [CrossRef]
- Vermeulen, F.; Le Camus, C.; Davies, J.C.; Bilton, D.; Milenkovic, D.; De Boeck, K. Variability of sweat chloride concentration in subjects with cystic fibrosis and G551D mutations. J. Cyst. Fibros. 2017, 16, 36–40. [Google Scholar] [CrossRef]
- Vermeulen, F.; Lebecque, P.; De Boeck, K.; Leal, T. Biological variability of the sweat chloride in diagnostic sweat tests: A retrospective analysis. J. Cyst. Fibros. 2017, 16, 30–35. [Google Scholar] [CrossRef]
- Cirilli, N.; Raia, V.; Rocco, I.; De Gregorio, F.; Tosco, A.; Salvadori, L.; Sepe, A.O.; Buzzetti, R.; Minicuci, N.; Castaldo, G. Intra-individual biological variation in sweat chloride concentrations in CF, CFTR dysfunction, and healthy pediatric subjects. Pediatr. Pulmonol. 2018, 53, 728–734. [Google Scholar] [CrossRef]
- Collaco, J.M.; Blackman, S.M.; Raraigh, K.S.; Corvol, H.; Rommens, J.M.; Pace, R.G.; Boelle, P.Y.; McGready, J.; Sosnay, P.R.; Strug, L.J.; et al. Sources of variation in sweat chloride measurements in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 1375–1382. [Google Scholar] [CrossRef]
- LeGrys, V.A.; Moon, T.C.; Laux, J.; Rock, M.J.; Accurso, F. Analytical and biological variation in repeated sweat chloride concentrations in clinical trials for CFTR modulator therapy. J. Cyst. Fibros. 2018, 17, 43–49. [Google Scholar] [CrossRef]
- Chin, T.; Williams, S.; Randhawa, I.; Nussbaum, E. Possible Eventual Positive Sweat Chloride Test Results in Children With CFTR-Related Metabolic Syndrome (CRMS). Chest J. 2011, 140, 909A. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Konstan, M.W.; Van Devanter, D.R.; Rowe, S.M.; Clancy, J.P.; Odem-Davis, K.; Skalland, M.; Mayer-Hamblett, N. Measuring the impact of CFTR modulation on sweat chloride in cystic fibrosis: Rationale and design of the CHEC-SC study. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- LeGrys, V.A.; Moon, T.C.; Laux, J.; Accurso, F.; Martiniano, S.A. A multicenter evaluation of sweat chloride concentration and variation in infants with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 190–193. [Google Scholar] [CrossRef]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551DCFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef]
- Fidler, M.C.; Beusmans, J.; Panorchan, P.; Van Goor, F. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J. Cyst. Fibros. 2017, 16, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Durmowicz, A.G.; Witzmann, K.A.; Rosebraugh, C.J.; Chowdhury, B.A. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: The ivacaftor experience. Chest 2013, 143, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Jones, A.M.; Webb, A.K.; Horsley, A.R. Sweat chloride is not a useful marker of clinical response to Ivacaftor. Thorax 2014, 69, 587–596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aalbers, B.L.; Hofland, R.W.; Bronsveld, I.; de Winter-de Groot, K.M.; Arets, H.G.M.; de Kiviet, A.C.; van Oirschot-van de Ven, M.M.M.; Kruijswijk, M.A.; Schotman, S.; Michel, S.; et al. Females with cystic fibrosis have a larger decrease in sweat chloride in response to lumacaftor/ivacaftor compared to males. J. Cyst. Fibros. 2021, 20, e7–e11. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Sagel, S.D.; Khan, U.; Heltshe, S.L.; Clancy, J.P.; Borowitz, D.; Gelfond, D.; Donaldson, S.H.; Moran, A.; Ratjen, F.; Van Dalfsen, J.M.; et al. Clinical Effectiveness of Lumacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del-CFTR. A Clinical Trial. Ann. Am. Thorac. Soc. 2021, 18, 75–83. [Google Scholar] [CrossRef]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbäurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef]
- Puchelle, E.; Gaillard, D.; Ploton, D.; Hinnrasky, J.; Fuchey, C.; Boutterin, M.C.; Jacquot, J.; Dreyer, D.; Pavirani, A.; Dalemans, W. Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am. J. Respir. Cell Mol. Biol. 1992, 7, 485–491. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Girodon, E.; Sands, D.; Stremmler, N.; Vavrova, V.; Deneuville, E.; Reix, P.; Bui, S.; Huet, F.; Lebourgeois, M.; et al. Clinical phenotype and genotype of children with borderline sweat test and abnormal nasal epithelial chloride transport. Am. J. Respir. Crit. Care Med. 2010, 182, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Yaakov, Y.; Omari, I.; Zaman, M.; Martin, C.R.; Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E.; Dasilva, D.; Sheth, S.; et al. Comparison of Nasal Potential Difference and Intestinal Current Measurements as Surrogate Markers for CFTR Function. J. Pediatr. Gastroenterol. Nutr. 2016, 63, e92–e97. [Google Scholar] [CrossRef] [PubMed]
- Kirilli, S.; Henry, T.; Wilschanski, M.; Fajac, I.; Davies, J.C.; Jais, J.P.; Sermet-Gaudelus, I.J. Insights into the variability of nasal potential difference, a biomarker of CFTR activity. J. Cyst. Fibros. 2020, 19, 620–626. [Google Scholar] [CrossRef]
- Vermeulen, F.; Proesmans, M.; Feyaerts, N.; De Boeck, K. Nasal potential measurements on the nasal floor and under the inferior turbinate: Does it matter? Pediatr. Pulmonol. 2011, 46, 145–152. [Google Scholar] [CrossRef]
- Vermeulen, F.; Proesmans, M.; Boon, M.; De Boeck, K. Improved repeatability of nasal potential difference with a larger surface catheter. J. Cyst. Fibros. 2015, 14, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.; Tümmler, B.; et al. Intestinal current measurements detect activation of mutant CFTR in patients with cystic fibrosis with the G551D mutation treated with ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, S.; Gonska, T.; Seydewitz, H.H.; Thomas, J.; Greiner, P.; Kuehr, J.; Brandis, M.; Eichler, I.; Rocha, H.; Lopes, A.; et al. CFTR Cl–channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004, 127, 1085–1095. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sermet-Gaudelus, I.; Nguyen-Khoa, T.; Hatton, A.; Hayes, K.; Pranke, I. Sweat Chloride Testing and Nasal Potential Difference (NPD) Are Primary Outcome Parameters in Treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. J. Pers. Med. 2021, 11, 729. https://doi.org/10.3390/jpm11080729
Sermet-Gaudelus I, Nguyen-Khoa T, Hatton A, Hayes K, Pranke I. Sweat Chloride Testing and Nasal Potential Difference (NPD) Are Primary Outcome Parameters in Treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. Journal of Personalized Medicine. 2021; 11(8):729. https://doi.org/10.3390/jpm11080729
Chicago/Turabian StyleSermet-Gaudelus, Isabelle, Thao Nguyen-Khoa, Aurélie Hatton, Kate Hayes, and Iwona Pranke. 2021. "Sweat Chloride Testing and Nasal Potential Difference (NPD) Are Primary Outcome Parameters in Treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators" Journal of Personalized Medicine 11, no. 8: 729. https://doi.org/10.3390/jpm11080729
APA StyleSermet-Gaudelus, I., Nguyen-Khoa, T., Hatton, A., Hayes, K., & Pranke, I. (2021). Sweat Chloride Testing and Nasal Potential Difference (NPD) Are Primary Outcome Parameters in Treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. Journal of Personalized Medicine, 11(8), 729. https://doi.org/10.3390/jpm11080729