
A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. CFTR Modulators
2.2. Cell Lines
2.3. Human Nasal Epithelia
2.4. PM Density Measurement
2.5. Peripheral Stability Measurement and Immunoblotting
2.6. Short-Circuit Current Measurement
2.7. Statistics
3. Results
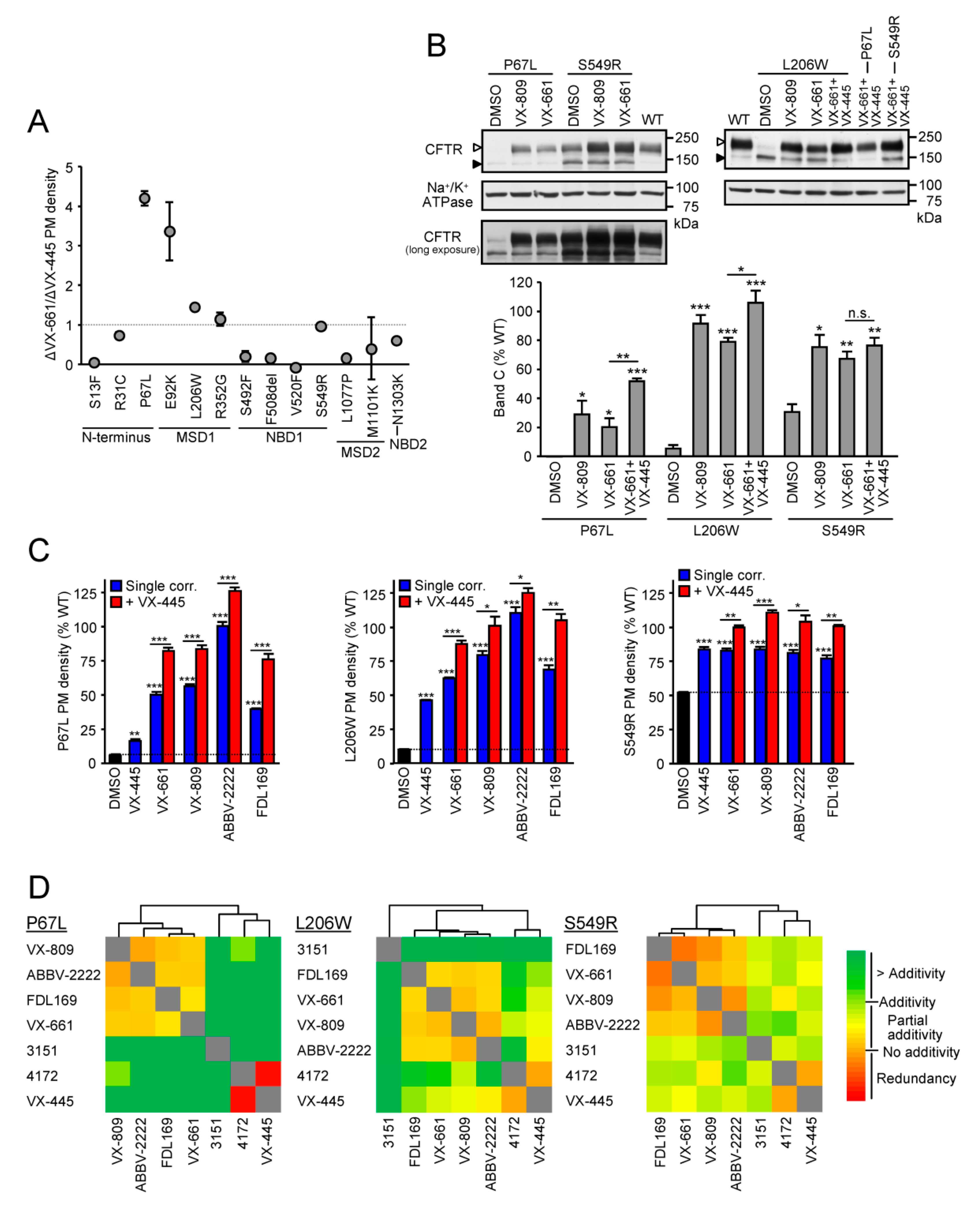
3.1. Identification of Mutants with High Responsiveness to Type I Correctors
3.2. Cooperative Correction of the Mature Protein and PM Expression by VX-661 + VX-445 Corrector Combination
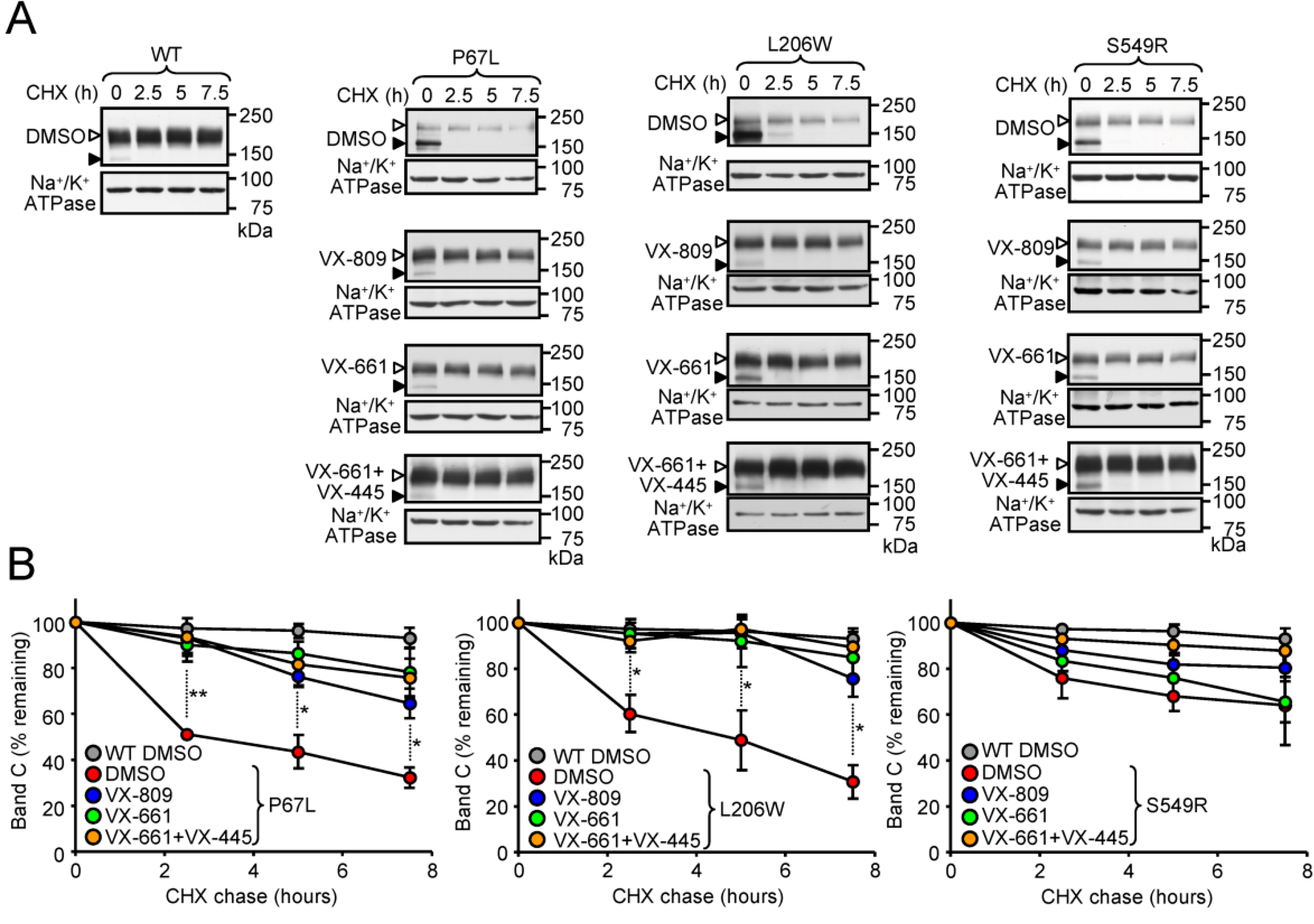
3.3. Correction of the Peripheral Stability Defect of P67L-, L206W- and S549R-CFTR
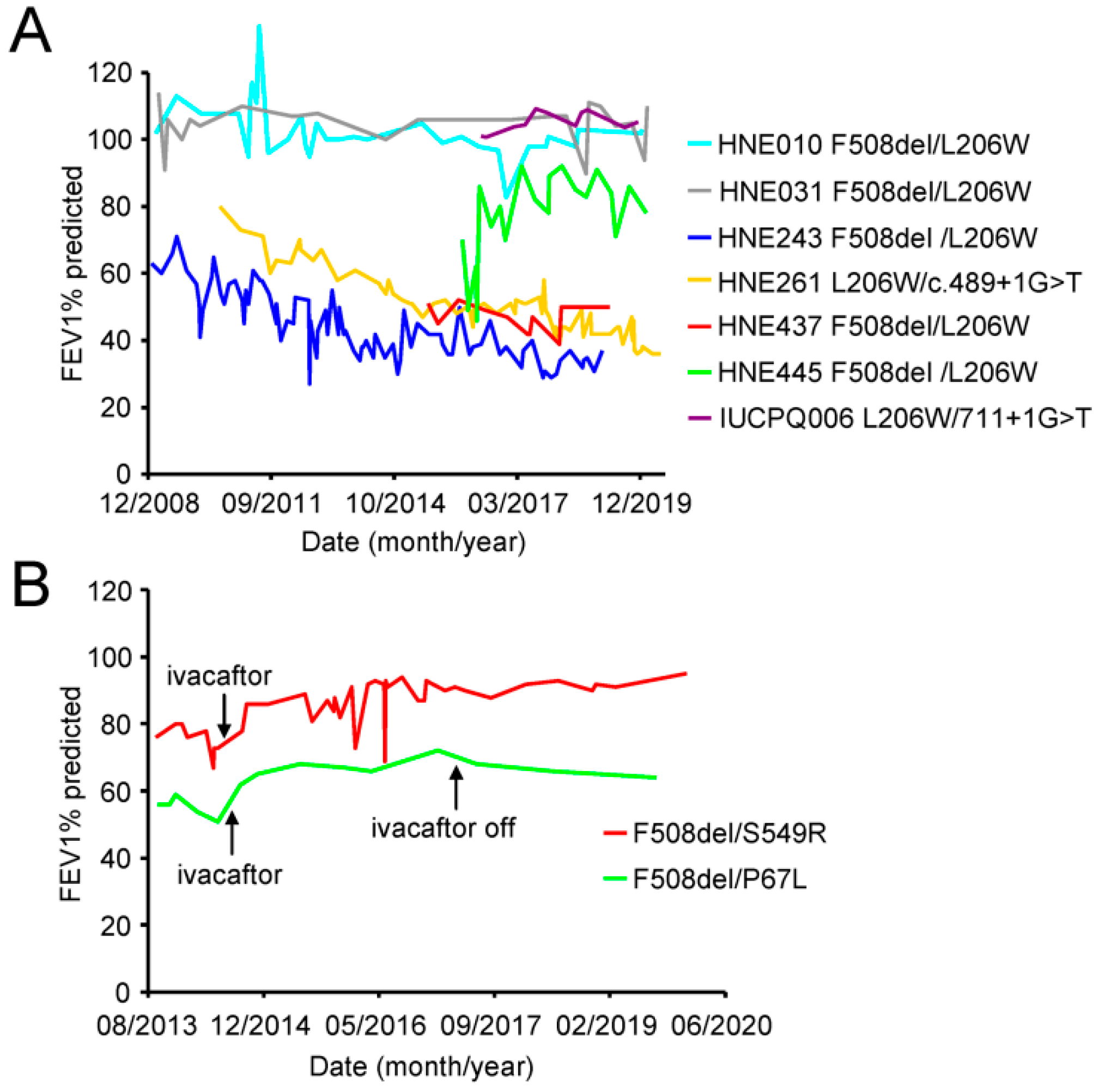
3.4. Some Patients with the L206W Mutation Exhibit Progressive Lung Function Decline
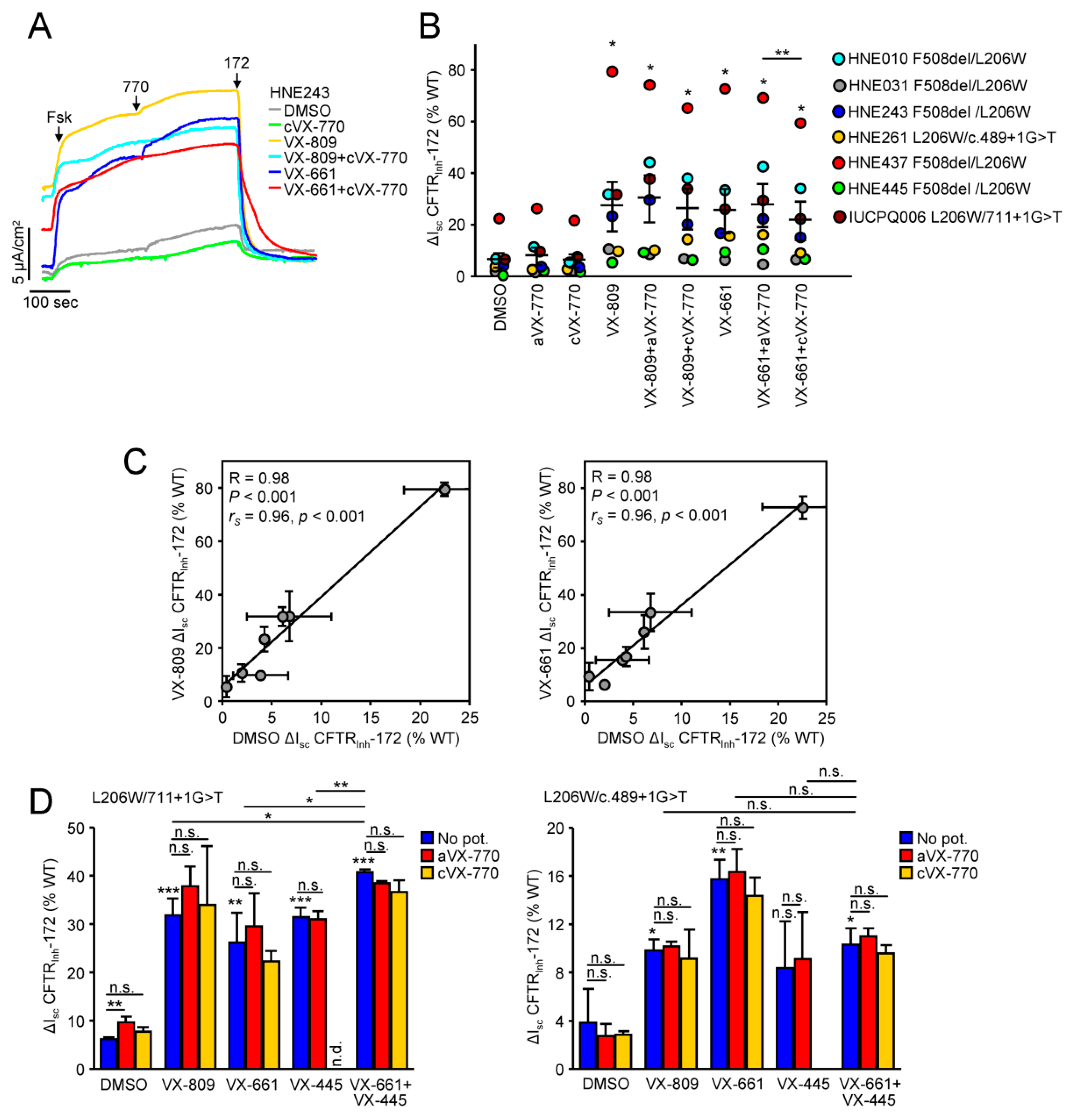
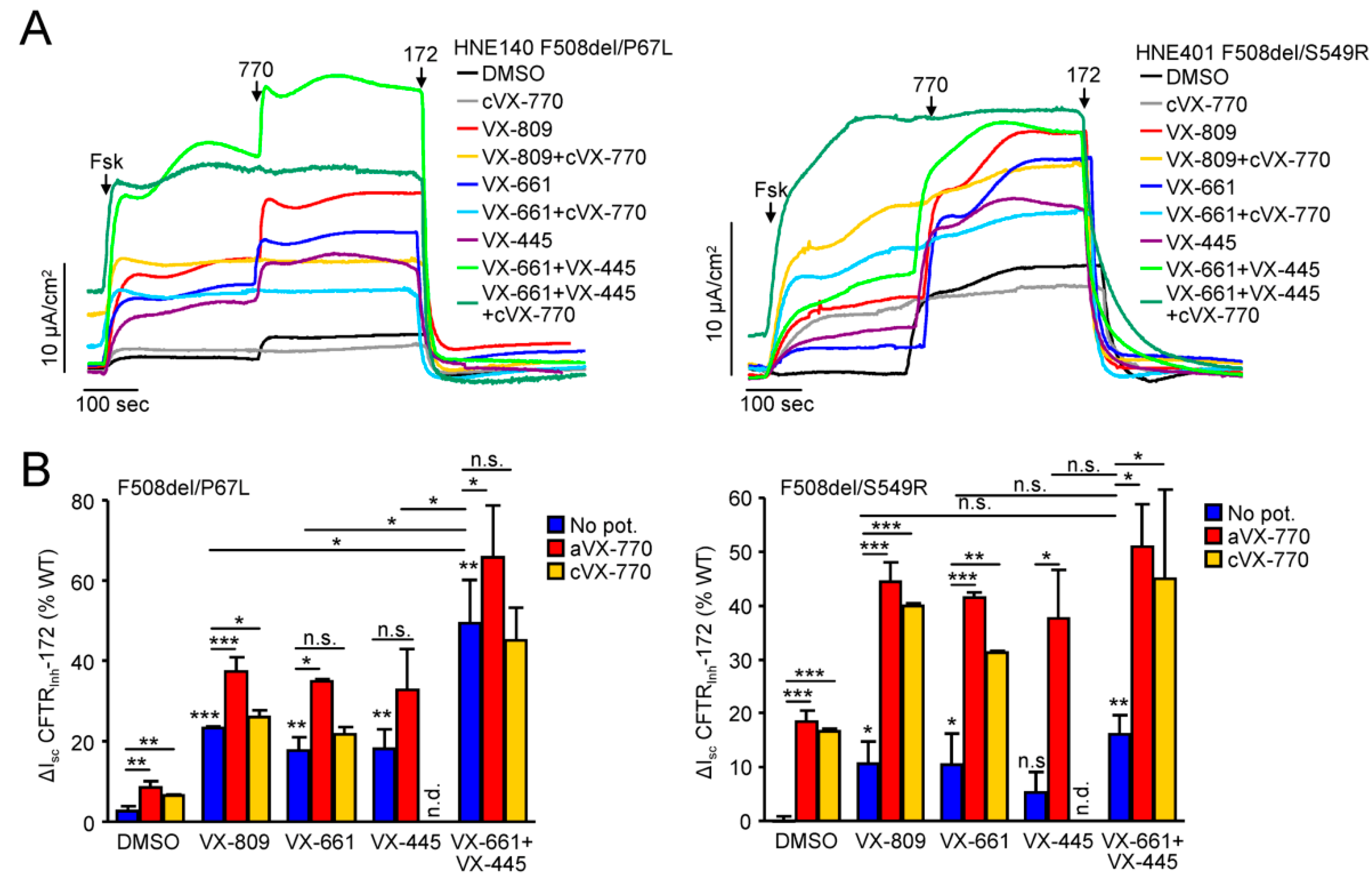
3.5. Functional Correction of P67L-, L206W- and S549R-CFTR in Human Nasal Epithelia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [Green Version]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.V.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef]
- Bompadre, S.G.; Li, M.; Hwang, T.C. Mechanism of G551D-CFTR (cystic fibrosis transmembrane conductance regulator) potentiation by a high affinity ATP analog. J. Biol. Chem. 2008, 283, 5364–5369. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M.; GOALe(2) Investigators. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, J.R. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Sharma, M.; Lukacs, G. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2004, 12, 17–25. [Google Scholar] [CrossRef]
- Du, K.; Lukacs, G.L. Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell. 2009, 20, 1903–1915. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.J.; Simon, A.; et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Molinski, S.V.; Shahani, V.M.; Subramanian, A.S.; MacKinnon, S.S.; Woollard, G.; Laforet, M.; Laselva, O.; Morayniss, L.D.; Bear, C.E.; Windemuth, A. Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins Struct. Funct. Bioinform. 2018, 86, 833–843. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the Major Cystic Fibrosis Mutant Interact through Membrane-Spanning Domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Bertozzi, S.M.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.; Ouyang, H.; Eckford, P.D.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J. Cyst. Fibros. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Bossard, F.; Goepp, J.; Verkman, A.S.; Galietta, L.; Hanrahan, J.W.; Lukacs, G.L. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell 2012, 23, 4188–4202. [Google Scholar] [CrossRef]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Avramescu, R.G.; Kai, Y.; Xu, H.; Bidaud-Meynard, A.; Schnúr, A.; Frenkiel, S.; Matouk, E.; Veit, G.; Lukacs, G.L. Mutation-specific downregulation of CFTR2 variants by gating potentiators. Hum. Mol. Genet. 2017, 26, 4873–4885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Sabusap, C.M.; Wang, W.; McNicholas, C.M.; Chung, W.J.; Fu, L.; Wen, H.; Mazur, M.; Kirk, K.L.; Collawn, J.F.; Hong, J.S.; et al. Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight 2016, 1, e86581. [Google Scholar] [CrossRef] [Green Version]
- Kleizen, B.; van Willigen, M.; Mijnders, M.; Peters, F.; Grudniewska, M.; Hillenaar, T.; Thomas, A.; Kooijman, L.; Peters, K.W.; Frizzell, R.; et al. Co-Translational Folding of the First Transmembrane Domain of ABC-Transporter CFTR is Supported by Assembly with the First Cytosolic Domain. J. Mol. Biol. 2021, 433, 166955. [Google Scholar] [CrossRef]
- Choi, M.; Park, S.; Yi, J.K.; Kwon, W.; Jang, S.; Kim, S.-Y.; Yu, W.; Kim, M.O.; Ryoo, Z.Y.; Choi, S.-K. Overexpression of hepatic serum amyloid A1 in mice increases IL-17-producing innate immune cells and decreases bone density. J. Biol. Chem. 2021, 296, 100595. [Google Scholar] [CrossRef]
- Bell, S.C.; Barry, P.J.; De Boeck, K.; Drevinek, P.; Elborn, J.S.; Plant, B.J.; Minić, P.; Van Braeckel, E.; Verhulst, S.; Muller, K.; et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J. Cyst. Fibros. 2019, 18, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsley, A.; Burr, L.; Kotsimbos, T.; Ledson, M.; Schwarz, C.; Simmonds, N.; O’Toole, T.; Kolodziej, A.; Ordoñez, C.; Bell, S. EPS3.06 Safety, pharmacokinetics and pharmacodynamics of the CFTR corrector FDL169. J. Cyst. Fibros. 2018, 17, S42. [Google Scholar] [CrossRef]
- Pomperada Sabusap, C.M.; Joshi, D.; Simhaev, L.; Oliver, K.E.; Senderowitz, H.; van Willigen, M.; Braakman, I.; Rab, A.; Sorscher, E.J.; Hong, J.S. The CFTR P67L variant reveals a key role for N-terminal lasso helices in channel folding, maturation, and pharmacologic rescue. J. Biol. Chem. 2021, 100598. [Google Scholar] [CrossRef] [PubMed]
- Clain, J.; Lehmann-Che, J.; Dugueperoux, I.; Arous, N.; Girodon, E.; Legendre, M.; Goossens, M.; Edelman, A.; de Braekeleer, M.; Teulon, J.; et al. Misprocessing of the CFTR protein leads to mild cystic fibrosis phenotype. Hum. Mutat. 2005, 25, 360–371. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, I.E.R.; Paquette, V.; Gosse, F.; George, S.; Chappe, F.; Chappe, V. Modeling cystic fibrosis disease progression in patients with the rare CFTR mutation P67L. J. Cyst. Fibros. 2017, 16, 335–341. [Google Scholar] [CrossRef]
- Dagan, A.; Cymberknoh, M.C.; Shteinberg, M.; Levine, H.; Vilozni, D.; Bezalel, Y.; Bar Aluma, B.-E.; Sarouk, I.; Ashkenazi, M.; Lavie, M.; et al. Ivacaftor for the p.Ser549Arg (S549R) gating mutation–The Israeli experience. Respir. Med. 2017, 131, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Burton, B.; Huang, C.-J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Winter-de Groot, K.M.; Berkers, G.; Marck-van der Wilt, R.E.P.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.K. Cytochrome P450 3A4 Induction: Lumacaftor versus Ivacaftor Potentially Resulting in Significantly Reduced Plasma Concentration of Ivacaftor. Drug Metab. Lett. 2018, 12, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Liu, Z.; Odom, L.V.; Kersh, L.; Guimbellot, J.S. CFTR Function and Clinical Response to Modulators Parallel Nasal Epithelial Organoid Swelling. Am. J. Physiol. Cell. Mol. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rabeh, W.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of Both NBD1 Energetics and Domain Interface Is Required to Restore ΔF508 CFTR Folding and Function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, J.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.; Thomas, P.J. Requirements for Efficient Correction of ΔF508 CFTR Revealed by Analyses of Evolved Sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef] [Green Version]
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021, 20, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Phuan, P.-W.; Haggie, P.M.; Tan, J.A.; Rivera, A.A.; Finkbeiner, W.E.; Nielson, D.W.; Thomas, M.M.; Janahi, I.A.; Verkman, A.S. CFTR modulator therapy for cystic fibrosis caused by the rare c.3700A>G mutation. J. Cyst. Fibros. 2021, 20, 452–459. [Google Scholar] [CrossRef]
- Laselva, O.; McCormack, J.; Bartlett, C.; Ip, W.; Gunawardena, T.; Ouyang, H.; Eckford, P.; Gonska, T.; Moraes, T.; Bear, C. Preclinical Studies of a Rare CF-Causing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J. Pers. Med. 2020, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Railean, V.; Duarte, A.; Amaral, M. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veit, G.; Velkov, T.; Xu, H.; Vadeboncoeur, N.; Bilodeau, L.; Matouk, E.; Lukacs, G.L. A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants. J. Pers. Med. 2021, 11, 643. https://doi.org/10.3390/jpm11070643
Veit G, Velkov T, Xu H, Vadeboncoeur N, Bilodeau L, Matouk E, Lukacs GL. A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants. Journal of Personalized Medicine. 2021; 11(7):643. https://doi.org/10.3390/jpm11070643
Chicago/Turabian StyleVeit, Guido, Tony Velkov, Haijin Xu, Nathalie Vadeboncoeur, Lara Bilodeau, Elias Matouk, and Gergely L. Lukacs. 2021. "A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants" Journal of Personalized Medicine 11, no. 7: 643. https://doi.org/10.3390/jpm11070643
APA StyleVeit, G., Velkov, T., Xu, H., Vadeboncoeur, N., Bilodeau, L., Matouk, E., & Lukacs, G. L. (2021). A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants. Journal of Personalized Medicine, 11(7), 643. https://doi.org/10.3390/jpm11070643