
Primary, Bilateral and Diffuse Renal Non-Hodgkin’s Lymphoma in a Young Woman Suffering from Turner Syndrome

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
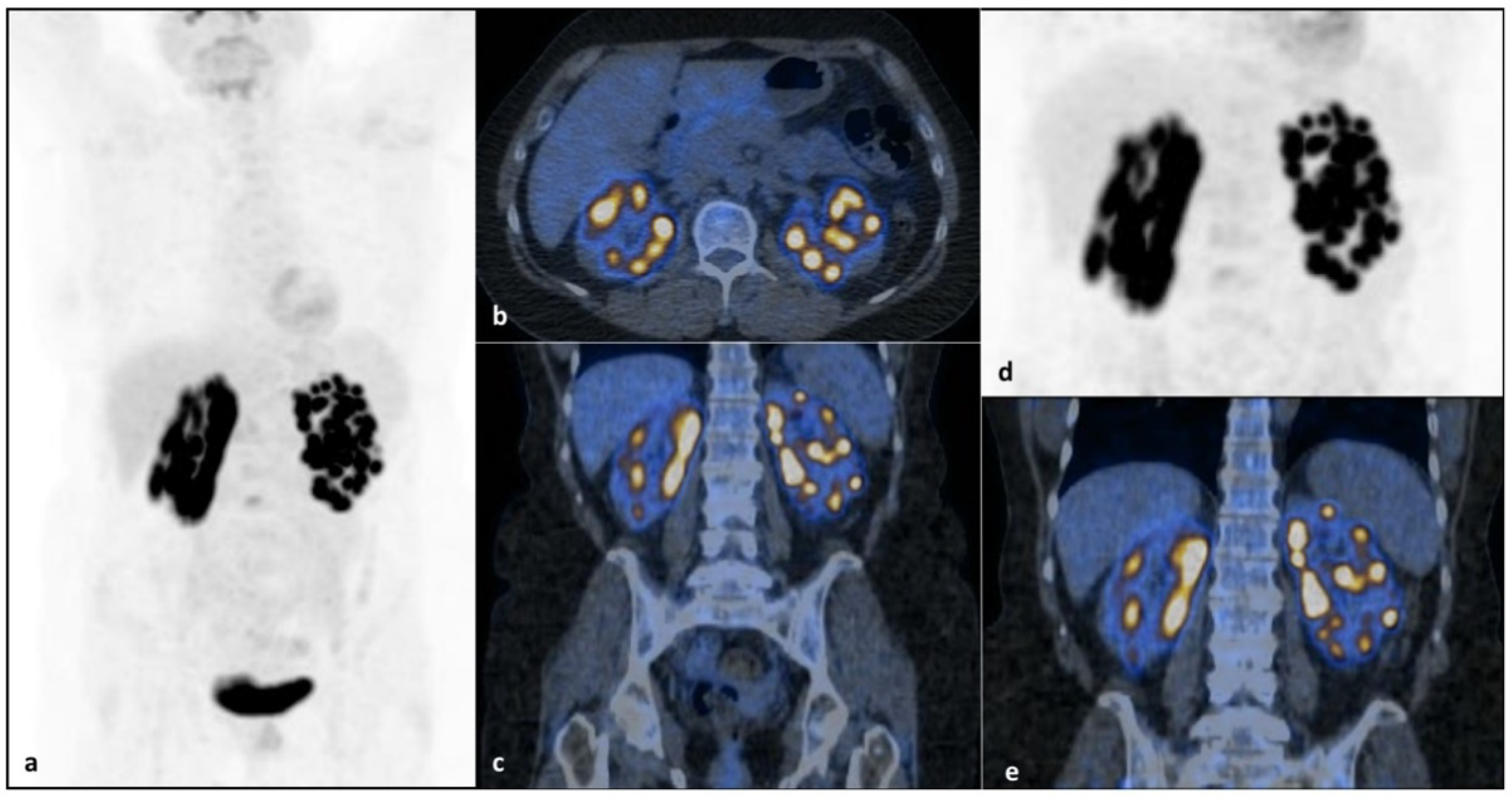
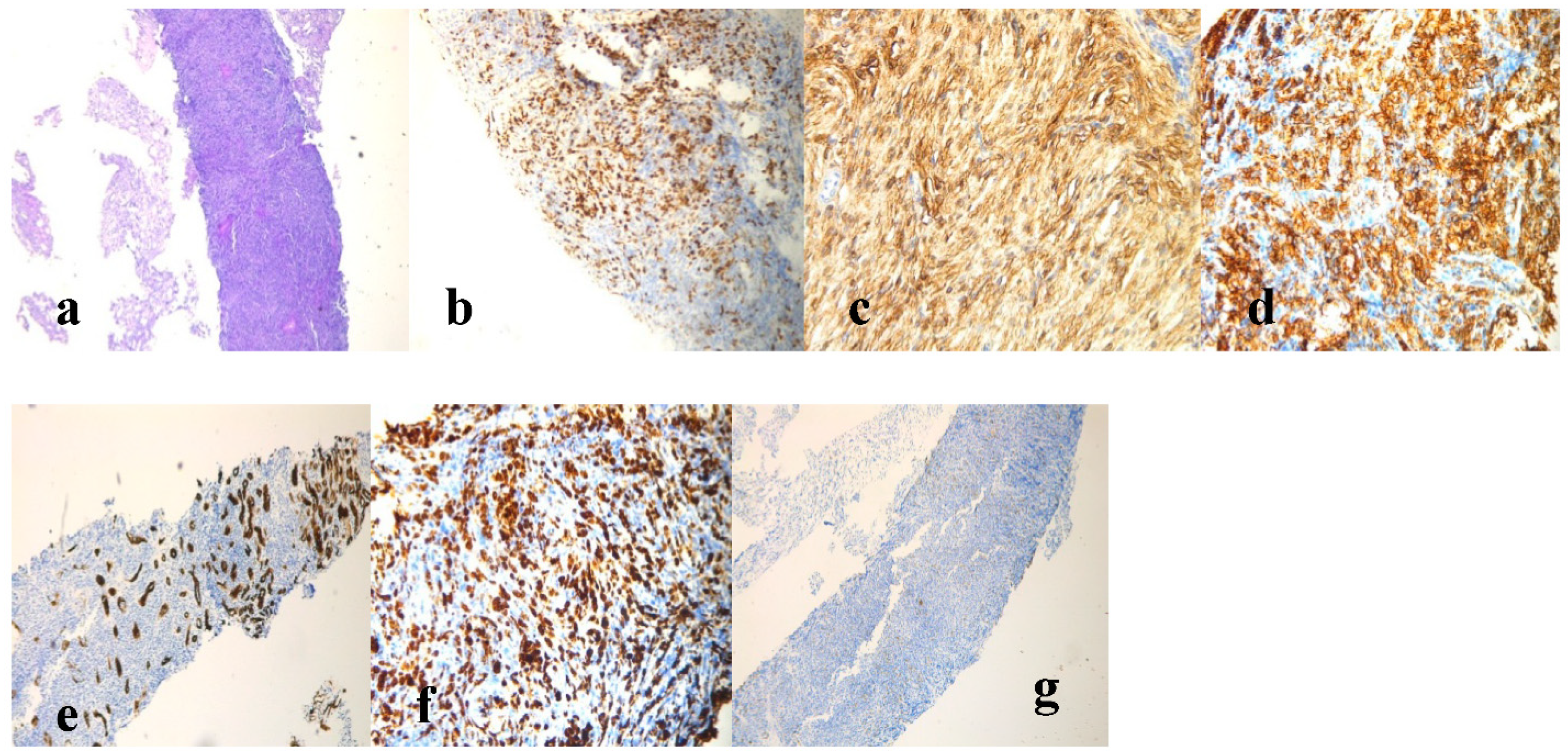
2. Case Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiang, H.; Zhong, W.; Gao, Q.; Bai, Y.; Wang, Z. Primary renal non-Hodgkin’s lymphoma: A clinicopathologic study of six cases and review of the literature. Int. J. Clin. Exp. Pathol. 2016, 9, 7436–7443. [Google Scholar]
- Chen, J.; Peng, J.; Zheng, Y.; Li, S.; Yang, P.; Wu, X.; Tian, H.; Liu, H.; Yang, S.; Wang, W.; et al. Primary renal lymphoma: A population-based study in the United States, 1980–2013. Sci. Rep. 2019, 9, 15125. [Google Scholar] [CrossRef] [Green Version]
- Taneja, A.; Kumar, V.; Chandra, A.B. Primary renal lymphoma: A population-based analysis using the SEER program (1973-2015). Eur. J. Haematol. 2020, 104, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Ladha, A.; Haider, G. Primary renal lymphoma. J. Coll. Physicians Surg. Pak. 2008, 18, 584–585. [Google Scholar]
- Chio, J.H.; Choi, G.B.; Shim, K.N.; Sung, S.H.; Han, W.S.; Baek, S.Y. Bilateral primary renal non-Hodgkin’s lymphoma presenting with acute renal failure. Successful treatment with systemic chemotherapy. Acta Haematol. 1997, 97, 231–235. [Google Scholar] [CrossRef]
- Cupisti, A.; Riccioni, R.; Carulli, G.; Paoletti, S.; Tognetti, A.; Meola, M.; Francesca, F.; Barsotti, G.; Petrini, M. Bilateral primary renal lymphoma treated by surgery and chemotherapy. Nephrol. Dial. Transplant. 2004, 19, 1629–1633. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Huang, Z.; Li, D.; Wang, Y. Enormous primary renal diffuse large B-cell lymphoma: A case report and literature review. J. Int. Med. Res. 2019, 47, 2728–2739. [Google Scholar] [CrossRef]
- Coggins, C.H. Renal failure in lymphoma. Kidney Int. 1980, 17, 847–855. [Google Scholar] [CrossRef] [Green Version]
- Belbaraka, R.; Elyoubi, M.B.; Boutayeb, S.; Errihani, H. Primary renal non-Hodgkin lymphoma: An unusual diagnosis for a renal mass. Indian J. Cancer 2011, 48, 255–256. [Google Scholar] [CrossRef]
- Ji, J.; Zöller, B.; Sundquist, J.; Sundquist, K. Risk of solid tumors and hematological malignancy in persons with Turner and Klinefelter syndromes: A national cohort study. Int. J. Cancer 2016, 139, 754–758. [Google Scholar] [CrossRef]
- Schoemaker, M.J.; Swerdlow, A.; Higgins, C.D.; Wright, A.F.; A Jacobs, P.; Schoemaker, M.J.; Swerdlow, A.; Higgins, C.D.; Wright, A.F.; A Jacobs, P. Cancer incidence in women with Turner syndrome in Great Britain: A national cohort study. Lancet Oncol. 2008, 9, 239–246. [Google Scholar] [CrossRef]
- Manola, K.N.; Sambani, C.; Karakasis, D.; Kalliakosta, G.; Harhalakis, N.; Papaioannou, M. Leukemias associated with Turner syndrome: Report of three cases and review of the literature. Leuk Res. 2008, 32, 481–486. [Google Scholar] [CrossRef] [PubMed]
- El-Mansoury, M.; Berntorp, K.; Bryman, I.; Hanson, C.; Innala, E.; Karlsson, A.; Landin-Wilhelmsen, K. Elevated liver enzymes in Turner syndrome during a 5-year follow-up study. Clin. Endocrinol. 2008, 68, 485–490. [Google Scholar] [CrossRef]
- Roulot, D. Liver involvement in Turner syndrome. Liver Int. 2013, 33, 24–30. [Google Scholar] [CrossRef]
- Fuchs, M.M.; Attenhofer, J.C.; Babovic-Vuksanovic, D.; Connolly, H.M.; Egbe, A. Long-Term Outcomes in Patients with Turner Syndrome: A 68-Year Follow-Up. J. Am. Heart Assoc. 2019, 8, e011501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. International Turner Syndrome Consensus Group. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 19, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Alliance, Australasian Leukaemia and Lymphoma Group; Eastern Cooperative Oncology Group; European Mantle Cell Lymphoma Consortium; Italian Lymphoma Foundation; et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef]
- Schmitz, N.; Zeynalova, S.; Nickelsen, M.; Kansara, R.; Villa, D.; Sehn, L.H.; Glass, B.; Scott, D.W.; Gascoyne, R.D.; Connors, J.M.; et al. CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J. Clin. Oncol. 2016, 34, 3150–3156. [Google Scholar] [CrossRef]
- Yim, S.K.; Yhim, H.Y.; Han, Y.H.; Jeon, S.Y.; Lee, N.R.; Song, E.K.; Jeong, H.J.; Kim, H.S.; Kwak, J.Y. Early risk stratification for diffuse large B-cell lymphoma integrating interim Deauville score and International Prognostic Index. Ann. Hematol. 2019, 98, 2739–2748. [Google Scholar] [CrossRef]
- Lauhan, C.; Schiff, N.; Gloude, N. Successful treatment of hepatosplenic T cell lymphoma in an adolescent with Turner syndrome usinifosfamide, carboplatin, and etoposide followed by allogeneic hematopoietic stem cell transplant. AuthoreaPrepr 2020. [Google Scholar] [CrossRef]
- Tilly, H.; Gomes da Silva, M.; Vitolo, U.; Jack, A.; Meignan, M.; Lopez-Guillermo, A.; Walewski, J.; André, M.; Johnson, P.W.; Pfreundschuh, M.; et al. ESMO Guidelines Committee. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v116–v125. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Networ Guidelines Version 4.2021 “B-Cell Lymphomas”. Available online: https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf (accessed on 5 June 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossini, B.; Skrypets, T.; Minoia, C.; Quinto, A.M.; Zaccaria, G.M.; Ferrari, C.; Maggialetti, N.; Mastrorosa, A.; Gatti, P.; Casiello, M.; et al. Primary, Bilateral and Diffuse Renal Non-Hodgkin’s Lymphoma in a Young Woman Suffering from Turner Syndrome. J. Pers. Med. 2021, 11, 644. https://doi.org/10.3390/jpm11070644
Rossini B, Skrypets T, Minoia C, Quinto AM, Zaccaria GM, Ferrari C, Maggialetti N, Mastrorosa A, Gatti P, Casiello M, et al. Primary, Bilateral and Diffuse Renal Non-Hodgkin’s Lymphoma in a Young Woman Suffering from Turner Syndrome. Journal of Personalized Medicine. 2021; 11(7):644. https://doi.org/10.3390/jpm11070644
Chicago/Turabian StyleRossini, Bernardo, Tetiana Skrypets, Carla Minoia, Angela Maria Quinto, Gian Maria Zaccaria, Cristina Ferrari, Nicola Maggialetti, Alessandro Mastrorosa, Pietro Gatti, Michela Casiello, and et al. 2021. "Primary, Bilateral and Diffuse Renal Non-Hodgkin’s Lymphoma in a Young Woman Suffering from Turner Syndrome" Journal of Personalized Medicine 11, no. 7: 644. https://doi.org/10.3390/jpm11070644
APA StyleRossini, B., Skrypets, T., Minoia, C., Quinto, A. M., Zaccaria, G. M., Ferrari, C., Maggialetti, N., Mastrorosa, A., Gatti, P., Casiello, M., Ciavarella, S., & Guarini, A. (2021). Primary, Bilateral and Diffuse Renal Non-Hodgkin’s Lymphoma in a Young Woman Suffering from Turner Syndrome. Journal of Personalized Medicine, 11(7), 644. https://doi.org/10.3390/jpm11070644