
Personalized Therapy and Liquid Biopsy—A Focus on Colorectal Cancer


Abstract
:1. Introduction
2. Liquid Biopsy (LB) and Targeted Approaches for Analysis
2.1. Types of Biopsies
2.1.1. Circulating Tumor Cells (CTCs)
Origin of CTCs
Epithelial to Mesenchymal Transition (EMT)
EMT, Stem Cells and Pathways
Mesenchymal-Epithelial Type (MET)
Characteristics of CTCs, Methods of Detection and Challenges
- Concentration gradient, Oncoquick®, because CTCs have a higher density than other cellular types [17]. While it was one of the first techniques that was developed, it is not seen as the most efficient. For example, in comparison with CellSearch® (Menarini Silicon Biosystems), in a cohort of 61 patients with cancers, only 23% of patients were found with at least 1 CTC with this technique while Cellsearch® (Menarini Silicon Biosystems), detected at least one CTC for more than 50% of patients [18].
- Filtration technique, Iset® [19], Rarecells Diagnostics SAS, since CTCs are larger than the other elements of the blood such as white cells. The main advantage of this technique relies on its independence from the presence of specific tumor cell markers. However, a drawback exists: some tumor cells are small and pass through filters [20].
2.1.2. Circulating Tumor DNA (ctDNA)
- Either necrosis or apoptosis of tumor cells in plasma;
- Or excreted from tumor cells within a vesicle called an exosome;
- Or contained within tumor cells.
2.2. Role of Liquid Biopsy in Adapted Cancer Treatment: Targeted Approaches for Analysis
2.2.1. Targeted Approaches with CTCs
2.2.2. Targeted Approaches with ctDNA
Droplet Digital PCR (ddPCR)
Beads, Emulsification, Amplification, and Magnetics (BEAMing)
3. Clinical Applications in Colorectal Cancer: Overview of the Current Literature
4. Contribution of the Omics to Personalize the Treatments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BEAMing | Beads: Emulsification, Amplification, and Magnetics |
| cfDNA | Circulating free DNA |
| CICs | Cancer initiating cells |
| CTCs | Circulating tumor cells |
| CRC | Colorectal cancer |
| ctDNA | Circulating tumor DNA |
| EMT | Epithelial to Mesenchymal Transition |
| EpCAM | Epithelial cell adhesion molecule |
| ECD | Extracellular domain |
| FDA | Food and Drug Administration |
| INCa | French National Cancer Institute |
| LB | Liquid Biopsy (ies) |
| MET | Mesenchymal-epithelial type |
| MRD | Minimal Residual Disease |
| TNF | Tumor necrosis factor |
| TRAIL | Apoptosis-inducing ligand |
References
- Karachaliou, N.; Mayo-de-las-Casas, C.; Molina-Vila, M.A.; Rosell, R. Real-time liquid biopsies become a reality in cancer treatment. Ann. Transl. Med. 2015, 3, 36. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4356857/ (accessed on 1 July 2021). [PubMed]
- Gingras, I.; Salgado, R.; Ignatiadis, M. Liquid biopsy: Will it be the ‘magic tool’ for monitoring response of solid tumors to anticancer therapies? Curr. Opin. Oncol. 2015, 27, 560–567. [Google Scholar] [CrossRef]
- Wu, J.; Hu, S.; Zhang, L.; Xin, J.; Sun, C.; Wang, L.; Ding, K.; Wang, B. Tumor circulome in the liquid biopsies for cancer diagnosis and prognosis. Theranostics 2020, 10, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Wills, B.; Gorse, E.; Lee, V. Role of liquid biopsies in colorectal cancer. Curr. Probl. Cancer 2018, 42, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T.R. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust. Med J. 1869, 14, 146–147. [Google Scholar]
- Mundy, G.R. Metastasis: Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and metastasis in colorectal cancer: Epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and β-catenin. Cells Tissues Organs. 2005, 179, 56–65. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Seki, T.; Okazaki, K. TGF-β during human colorectal carcinogenesis: The shift from epithelial to mesenchymal signaling. Inflammopharmacology 2006, 14, 198–203. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Steven, A.E.; Timothy, J.Y.; Natasha, G.D.; R. Daniel, B. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 2012, 142, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.A.; Cooper, B.W.; Lazarus, H.M.; Mackay, W.; Moss, T.J.; Ciobanu, N.; Tallman, M.S.; Kennedy, M.J.; Davidson, N.E.; Sweet, D.; et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993, 82, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Müller, V.; Stahmann, N.; Riethdorf, S.; Rau, T.; Zabel, T.; Goetz, A.; Jänicke, F.; Pantel, K. Circulating tumor cells in breast cancer: Correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin. Cancer Res. 2005, 11, 3678–3685. [Google Scholar] [CrossRef] [Green Version]
- Balic, M.; Dandachi, N.; Hofmann, G.; Samonigg, H.; Loibner, H.; Obwaller, A.; van der Kooi, A.; Tibbe, A.G.J.; Doyle, G.V.; Terstappen, L.W.M.M.; et al. Bauernhofer, T. Comparison of two methods for enumerating circulating tumor cells in carcinoma patients. Cytom. B Clin. Cytom. 2005, 68, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schütze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by Size of Epithelial Tumor Cells: A New Method for the Immunomorphological and Molecular Characterization of Circulating Tumor Cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Farace, F.; Massard, C.; Vimond, N.; Drusch, F.; Jacques, N.; Billiot, F.; Laplanche, A.; Chauchereau, A.; Lacroix, L.; Planchard, D.; et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br. J. Cancer 2011, 105, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.-V.; Jen, C.-P. Impedance detection integrated with dielectrophoresis enrichment platform for lung circulating tumor cells in a microfluidic channel. Biosens Bioelectron. 2018, 121, 10–18. [Google Scholar] [CrossRef]
- Le Du, F.; Fujii, T.; Kida, K.; Davis, D.W.; Park, M.; Liu, D.D.; Wu, W.; Chavez-MacGregor, M.; Barcenas, C.H.; Valero, V.; et al. EpCAM-independent isolation of circulating tumor cells with epithelial-to-mesenchymal transition and cancer stem cell phenotypes using ApoStream® in patients with breast cancer treated with primary systemic therapy. PLoS ONE 2020, 15, e0229903. [Google Scholar] [CrossRef]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W.W.M. Circulating Tumor Cells at Each Follow-up Time Point during Therapy of Metastatic Breast Cancer Patients Predict Progression-Free and Overall Survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Kularatne, S.A.; Kalli, K.R.; Prendergast, F.G.; Amato., R.J.; Klee, G.G.; Hartmann, L.C.; Low, P.S. Quantitation of circulating tumor cells in blood samples from ovarian and prostate cancer patients using tumor-specific fluorescent ligands. Int. J. Cancer 2008, 123, 1968–1973. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Piva, F.; Marinelli, O.; Maggi, F.; Bianchi, F.; Bittoni, A.; Berardi, R.; Giampieri, R.; et al. Expression Profiling of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma Patients: Biomarkers Predicting Overall Survival. Front. Oncol. 2019, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Labib, M.; Kelley, S.O. Circulating tumor cell profiling for precision oncology. Mol. Oncol. 2021, 15, 1622–1646. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. [Nuclear Acids in human blood plasma.]. C R Seances Soc Biol Fil. 1948, 142, 241–243. [Google Scholar]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2018, 42, 95–106. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano1, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.C.; Robinson, P.S.; Wagner, C.; O’Shannessy, D.J. The ParsortixTM Cell Separation System—A versatile liquid biopsy platform. Cytom. Part A 2018, 93, 1234–1239. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaill, V.; Zheng, Z.; et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, W.; Wang, Z.; Tang, Y.; Deng, Y.; Xu, L.; Tian, J.; Shi, Q. Ex vivo expansion of circulating lung tumor cells based on one-step microfluidics-based immunomagnetic isolation. Analyst 2016, 141, 3621–3625. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.; Song, H.; Sohn, K.Y.; Jeon, M.; Han, K.-H. Microfluidic technologies for circulating tumor cell isolation. Analyst 2018, 143, 2936–2970. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Dubash, T.D.; Edd, J.F.; Jewett, M.K.; Garre, S.G.; Karabacak, N.M.; Rabe, D.C.; Mutlu, B.R.; Walsh, J.R.; Kapur, R.; et al. Ultrahigh-throughput magnetic sorting of large blood volumes for epitope-agnostic isolation of circulating tumor cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16839–16847. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Wayne, E.; Rana, K.; Schaffer, C.B.; King, M.R. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc. Natl. Acad. Sci. USA 2014, 111, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pellè, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.M.; Spellman, P.T.; Gray, J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr. Opin. Genet. Dev. 2017, 42, 14–21. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- La Situation du Cancer en France en 2012. In Collection Etat des Lieux et des Connaissances; Ouvrage Collectif édité par l’INCa: Boulogne-Billancourt, France, 2013.
- Xu, J.-M.; Liu, X.-J.; Ge, F.-J.; Lin, L.; Wang, Y.; Sharma, M.R.; Xu, J.-M.; Liu, X.-J.; Ge, F.-J.; Lin, L.; et al. KRAS mutations in tumor tissue and plasma by different assays predict survival of patients with metastatic colorectal cancer. J. Exp. Clin. Cancer Res. 2014, 33, 104. [Google Scholar] [CrossRef]
- Yamanaka, T.; Oki, E.; Yamazaki, K.; Yamaguchi, K.; Muro, K.; Uetake, H.; Sato, T.; Nishina, T.; Ikeda, M.; Kato, T.; et al. 12-Gene Recurrence Score Assay Stratifies the Recurrence Risk in Stage II/III Colon Cancer With Surgery Alone: The sunrise Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2906–2913. [Google Scholar] [CrossRef]
- Taieb, J.; Jung, A.; Sartore-Bianchi, A.; Peeters, M.; Seligmann, J.; Zaanan, A.; Burdon, P.; Montagut, C.; Laurent-Puig, P. The Evolving Biomarker Landscape for Treatment Selection in Metastatic Colorectal Cancer. Drugs 2019, 79, 1375–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Lenz, H.-J.; Köhne, C.-H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.J.M.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Amatu, A.; Porcu, L.; Ghezzi, S.; Lonardi, S.; Leone, F.; Bergamo, F.; Fenocchio, E.; Martinelli, E.; Borelli, B.; et al. HER2 Positivity Predicts Unresponsiveness to EGFR-Targeted Treatment in Metastatic Colorectal Cancer. Oncologist 2019, 24, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- French, A.J.; Sargent, D.J.; Burgart, L.J.; Foster, N.R.; Kabat, B.F.; Goldberg, R.; Shepherd, L.; Windschitl, H.E.; Thibodeau, S.N. Prognostic Significance of Defective Mismatch Repair and BRAF V600E in Patients with Colon Cancer. Clin. Cancer Res. 2008, 14, 3408–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Eric, V.C.; Ray, M.; Hill, A. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [Green Version]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients with RAS and BRAF Wild-Type Metastatic Colorectal Cancer with Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.-C.; Kiavue, N.; Ychou, M.; Cabel, L.; Stern, M.-H.; Madic, J.; Saliou, A.; Rampanou, A.; Decraene, C.; Bouché, O.; et al. Circulating Tumor Cells and Circulating Tumor DNA Detection in Potentially Resectable Metastatic Colorectal Cancer: A Prospective Ancillary Study to the Unicancer Prodige-14 Trial. Cells 2019, 8, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindler, K.-L.G.; Pallisgaard, N.; Appelt, A.L.; Andersen, R.F.; Schou, J.V.; Nielsen, D.; Pfeiffer, P.; Yilmaz, M.; Johansen, J.S.; Hoegdall, E.V.; et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor therapy. Eur. J. Cancer 2015, 51, 2678–2685. [Google Scholar] [CrossRef]
- Chen, E.X.; Jonker, D.J.; Loree, J.M.; Kennecke, H.F.; Berry, S.R.; Couture, F.; Ahmad, C.E.; Goffin, J.R.; Kavan, P.; Harb, M.; et al. Effect of Combined Immune Checkpoint Inhibition vs Best Supportive Care Alone in Patients With Advanced Colorectal Cancer: The Canadian Cancer Trials Group CO.26 Study. JAMA Oncol. 2020, 6, 831–838. [Google Scholar] [CrossRef]
- Khan, K.; Rata, M.; Cunningham, D.; Koh, D.-M.; Tunariu, N.; Hahne, J.C.; Hedayat, S.; Marchetti, S.; Lampis, A.; Damavandi, M.D.; et al. Functional imaging and circulating biomarkers of response to regorafenib in treatment-refractory metastatic colorectal cancer patients in a prospective phase II study. Gut 2018, 67, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Elez, E.; Chianese, C.; Sanz-García, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.M.; Caratù, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Impact of circulating tumor DNA mutant allele fraction on prognosis in RAS-mutant metastatic colorectal cancer. Mol. Oncol. 2019, 13, 1827–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.; Price, T.; Boedigheimer, M.; Kim, T.W.; Ruff, P.; Gibbs, P.; Thomas, A.; Demonty, G.; Hool, K.; Ang, A.; et al. Evaluation of Emergent Mutations in Circulating Cell-Free DNA and Clinical Outcomes in Patients with Metastatic Colorectal Cancer Treated with Panitumumab in the ASPECCT Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Appelt, A.L.; Andersen, R.F.; Lindebjerg, J.; Jakobsen, A. Prognostic Value of Serum NPY Hypermethylation in Neoadjuvant Chemoradiotherapy for Rectal Cancer: Secondary Analysis of a Randomized Trial. Am. J. Clin. Oncol. 2020, 43, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Janowski, E.; Timofeeva, O.; Chasovskikh, S.; Goldberg, M.; Kim, A.; Banovac, F.; Pang, D.; Dritschilo, A.; Unger, K. Yttrium-90 radioembolization for colorectal cancer liver metastases in KRAS wild-type and mutant patients: Clinical and ccfDNA studies. Oncol. Rep. 2017, 37, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Holm, M.; Andersson, E.; Osterlund, E.; Ovissi, A.; Soveri, L.-M.; Anttonen, A.-K.; Kytölä, S.; Aittomäki, K.; Osterlund, P.; Ristimäki, A.; et al. Detection of KRAS mutations in liquid biopsies from metastatic colorectal cancer patients using droplet digital PCR, Idylla, and next generation sequencing. PLoS ONE 2020, 15, e0239819. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7688175/ (accessed on 1 July 2021). [PubMed]
- Ning, Y.; Zhang, W.; Hanna, D.L.; Yang, D.; Okazaki, S.; Berger, M.D.; Miyamoto, Y.; Suenaga, M.; Schirripa, M.; El-Khoueiry, A.; et al. Clinical relevance of EMT and stem-like gene expression in circulating tumor cells of metastatic colorectal cancer patients. Pharm. J. 2018, 18, 29–34. [Google Scholar] [CrossRef]
- Matikas, A.; Souglakos, J.; Katsaounis, P.; Kotsakis, A.; Kouroupakis, P.; Pantazopoulos, N.; Kentepozidis, N.; Nikolaidi, A.; Messaritakis, I.; Tzovara, I.; et al. MINOAS: A Single-arm Translational Phase II Trial of FOLFIRI Plus Aflibercept as First-line Therapy in Unresectable, Metastatic Colorectal Cancer. Target Oncol. 2019, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Soster, M.; Juris, R.; Bonacchi, S.; Genovese, D.; Montalti, M.; Rampazzo, E.; Zaccheroni, N.; Garagnani, P.; Bussolino, F.; Prodi, L.; et al. Targeted dual-color silica nanoparticles provide univocal identification of micrometastases in preclinical models of colorectal cancer. Int. J. Nanomed. 2012, 7, 4797–4807. [Google Scholar]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.-M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef]
- Bölke, E.; Orth, K.; Gerber, P.A.; Lammering, G.; Mota, R.; Peiper, M.; Matuschek, C.; Budach, W.; Rusnak, E.; Shaikh, S.; et al. Gene expression of circulating tumour cells and its correlation with tumour stage in breast cancer patients. Eur. J. Med. Res. 2009, 14, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusaka, S.; Chìn, K.; Ogura, M.; Suenaga, M.; Shinozaki, E.; Mishima, Y.; Terui, Y.; Mizunuma, N.; Hatake, K. Circulating tumor cells as a surrogate marker for determining response to chemotherapy in patients with advanced gastric cancer. Cancer Sci. 2010, 101, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Koutsilieris, M. Diagnostic value of circulating tumor cell detection in bladder and urothelial cancer: Systematic review and meta-analysis. BMC Cancer 2011, 11, 336. [Google Scholar] [CrossRef] [Green Version]
- Somlo, G.; Lau, S.K.; Frankel, P.; Hsieh, H.B.; Liu, X.; Yang, L.; Krivacic, R.; Bruce, R.H. Multiple biomarker expression on circulating tumor cells in comparison to tumor tissues from primary and metastatic sites in patients with locally advanced/inflammatory, and stage IV breast cancer, using a novel detection technology. Breast Cancer Res. Treat. 2011, 128, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Pons, M.; Cruz-Correa, M. Colorectal Cancer Biomarkers: Where Are We Now? BioMed Res. Int. 2015, 2015, 149014. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4461726/ (accessed on 1 July 2021). [CrossRef] [Green Version]
- Malapelle, U.; Pisapia, P.; Sgariglia, R.; Vigliar, E.; Biglietto, M.; Carlomagno, C.; Giuffrè, G.; Bellevicine, C.; Troncone, G. Less frequently mutated genes in colorectal cancer: Evidences from next-generation sequencing of 653 routine cases. J. Clin. Pathol. 2016, 69, 767–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pira, G.; Uva, P.; Scanu, A.M.; Rocca, P.C.; Murgia, L.; Uleri, E.; Piu, C.; Porcu, A.; Carru, C.; Manca, A.; et al. Landscape of transcriptome variations uncovering known and novel driver events in colorectal carcinoma. Sci. Rep. 2020, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Cifani, P.; Kentsis, A. Towards comprehensive and quantitative proteomics for diagnosis and therapy of human disease. Proteomics 2017, 17, 1600079. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5243792/ (accessed on 1 July 2021). [CrossRef] [Green Version]
- Ku, X.; Xu, Y.; Cai, C.; Yang, Y.; Cui, L.; Yan, W. In-Depth Characterization of Mass Spectrometry-Based Proteomic Profiles Revealed Novel Signature Proteins Associated with Liver Metastatic Colorectal Cancers. Anal. Cell Pathol. 2019, 2019, 7653230. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Chaver, P. Proteomics for discovery of candidate colorectal cancer biomarkers. World J. Gastroenterol. 2014, 20, 3804. [Google Scholar] [CrossRef]
- Madunić, K.; Zhang, T.; Mayboroda, O.A.; Holst, S.; Stavenhagen, K.; Jin, C.; Karlsson, N.G.; Lageveen-Kammeijer, G.S.M.; Wuhrer, M. Colorectal cancer cell lines show striking diversity of their O-glycome reflecting the cellular differentiation phenotype. Cell. Mol. Life Sci. 2020, 78, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Fu, Y.; Huang, Z.; Liu, Y.; Liu, B.-F.; Cheng, L.; Liu, X. A comprehensive analysis of subclass-specific IgG glycosylation in colorectal cancer progression by nanoLC-MS/MS. Analyst 2020, 145, 3136–3147. [Google Scholar] [CrossRef]

| CTCs | ctDNA | |
|---|---|---|
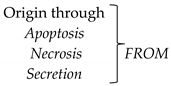 | Tumor cells Metastatic cells | Tumor cells Metastatic cells CTCs |
| Mode of isolation for further analysis | -Direct visualization -Physical separation -Biological separation/immunoaffinity | Nucleic acid samples pre-kit |
| Analysis capability | -Phenotypic studies -Proteomics -Genomics -Transcriptomics | -Genetics -Epigenetics |
| Technologies available | -FISH -Flow-cytometry in vitro/in vivo studies -RT-PCR | -NGS -RT -PCR -ddPCR |
| Pros | -Wide analysis -Functional analysis possible -Molecular analysis at both cellular and sub-cellular levels | -Abundant -Encompasses genetic heterogeneity |
| Cons | -Rare and fragile -False-negative and false-positive results | -Pre analytical conditions not standardized |
| A step forward personalized treatments | Biological and immunological test: -drug sensitivity and drug resistance prediction -CTCs clusters-xenograft models | Molecular testing with targetable genomic alterations: gene expression and mutations, genomic signatures |
| Clinical Trial | Inclusion Criteria and Type of Intervention | Main Outcome | Methodology for ctDNA Analysis | Results |
|---|---|---|---|---|
| NCT02994888 [52] | Patients with RAS wild-type (WT), refractory metastatic CRC (mCRC) | Mechanisms of resistance/response to anti-EGFR therapies | -ddPCR and ultra-deep next generation sequencing (NGS) | Primary and acquired resistance to anti-EGFR: polyclonal, and observed in both tissue and plasma samples |
| NCT02296203 [53] | Cetuximab plus irinotecan as third-line treatment for patients with RAS and BRAF WT mCRC who were initially sensitive to and then resistant to first-line irinotecan- and cetuximab-based therapy | Percentage of patients achieving a decrease ≥ 30% in the sum of the longest diameters of target lesions | -ddPCR and ultra-deep next generation sequencing (NGS) | Rechallenge by cetuximab plus irinotecan is an active option for patients with RAS and BRAF wild-type metastatic colorectal cancer who have acquired resistance to first-line irinotecan- and cetuximab-based therapy but with RAS and BRAF wild-type circulating tumor DNA at the time of rechallengeLB leads to track molecular events in (ctDNA) through the different lines of chemotherapy |
| NCT01442935 [54] | Metastatic disease with synchronous or metachronous (>3 months after diagnosis of the primary tumor) non resectable liver metastasis (LM) | Compare resection rates (R0 or R1) for hepatic metastases | -CTC detected with Cellsearch® -KRAS ctDNA (droplet digital polymerase chain reaction (PCR)) levels were assessed at inclusion, after 4 weeks of therapy and before LM surgery. | -CTC detection at 4 weeks (≥1 or ≥3 CTC) was not significantly associated with the eventual R0/R1 resection of LM (p = 0.06) -Persistently detectable KRAS ctDNA at 4 weeks after neoadjuvant chemotherapy was associated with a lower R0/R1 liver metastasis (LM) resection rate Possible selection of eligible patients to thanks to ctDNA |
| VEK nr. H-KA-20060094 [55] | Patients resistant to 5-FU, oxaliplatin and irinotecan and treated with 3rd line irinotecan and cetuximab | Clinical value of KRAS mutations when detected in plasma compared to tumor in patients from mCRC prior to anti-EGFR therapy | RT-PCR | KRAS detection in archival tumor tissue showed no correlation to survival, whereas plasma KRAS status remained a strong predictive and prognostic factor in multivariate analysis |
| NCT02870920. [56] | -Patients with CRC receiving all available standard systemic therapies (fluoropyrimidines, oxaliplatin, irinotecan, and bevacizumab if appropriate; cetuximab or panitumumab if RAS wild-type tumors; regorafenib if available)-Randomized in either Combined Immune Checkpoint Inhibition vs. Best Supportive Care Alone | Overall survival (OS) | cfDNA collected prior to study therapy, at 8 weeks, and at the time of disease progression, with GuardantOMNI next generation sequencing 2.15 Mb, 500-gene panel (Guardant Health, Inc) | Patients with Microsatellite Stable (MSS) status, had significantly improved OS with durvalumab and tremelimumab (p = 0.02). Patients who were MSS with plasma Tumor Mutation Burden (TMB) of 28 variants per megabase or more (21% of MSS patients) had the greatest OS benefit (p = 0.004). |
| NCT03010722 [57] | Patients with chemorefractory RAS mutant metastatic CRC received Regorafenib | Overall survival (OS) and Progression Free Survival (PFS) | clonal RAS mutations by digital-droplet PCR. | RAS mutant clones decay in ctDNA after 8 weeks of treatment was associated with better PFS (p = 0.01) and OS (p = 0.06) => ctDNA may predict duration of anti-angiogenic response to regorafenib |
| NCT01704703 [58] | RAS-mutated patients with nonresectable metastases from CRC | Overall survival (OS) and Progression Free Survival (PFS) | RAS testing in ctDNA using BEAMing before first- and/or second-line treatment | RAS mutant allele fraction (MAFs) is an independent prognostic factor in both OS (p = 0.006) and first-line PFS (p = 0.049). |
| NCT01001377 [59] | -Patients with chemo-refractory wild-type KRAS exon 2 mCRC- randomized 1:1 to receive either panitumumab or cetuximab | Overall survival (OS) | -Plasma samples analyzed at baseline and safety follow-up (SFU) by a next-generation sequencing-based approach for extended RAS mutant allele frequency (MAF)-Mutational status of EGFR pathway genes was also examined | Despite observed trend of higher RAS MAF correlating with worse outcomes, baseline extended RAS mutations did not preclude clinical response to panitumumab. Even though baseline mutations in EGFR pathway genes were associated with shorter OS, the prognostic model analyzing mutations as a continuous variable showed that patients who were mutant for EGFR pathway genes but with a low MAF may still derive clinical benefit from panitumumab. |
| Appelt et al., 2020 [60] | Patients with MRI-staged T3-4N0-2M0 rectal cancer treated by neoadjuvant chemoradiotherapy | Overall survival (OS) and the rate of distant metastases were compared between meth-ctDNA (hypermethylation of the neuropeptide Y gene-ctDNA) positive and negative patients | ddPCR | Patients with meth-ctDNA had significantly worse 5-year OS |
| Janowski et al., 2017 [61] | Patients benefited from resin-based yttrium-90 (90Y) radioembolization for unresectable liver metastasis from CRC | Overall survival (OS) and DNA fragmentation index (FI) quantification | circulating cell-free DNA (ccfDNA) concentration and fragmentation index (FI) were measured using quantitative PCR and atomic-force microscopy (AFM) | In the WT and KRAS mutant patients, DNA FI was reduced after treatment. This reduction was associated with an improved OS (p = 0.046). Analysis by AFM of paired pre- and post-treatment samples from KRAS mutant and WT patients revealed significant decrease in fragment size in the WT patients (p = 0.013). |
| NCT01531595 [62] | Metastatic colorectal cancer (mCRC) patients with a known KRAS mutation in their primary tumor underwent oncological treatment with bevacizumab in combination with alternating capecitabine and oxaliplatin or irinotecan | Compare multiple methods for measuring KRAS mutations in periodically collected liquid biopsies | Plasma ddPCR KRAS mutation allele frequency (MAF) versus Plasma real-time PCR based molecular testing system (IdyllaTM ctKRAS cartridge) versusPlasma Next-generation sequencing (NGS) [Ion AmpliSeq Hotspot Panel v2, which surveys the hotspot regions of 50 oncogenes and tumor suppressor genes (Thermo Fisher Scientific, Waltham, MD, USA)] | ddPCR and IdyllaTM are equally efficient for the detection of KRAS mutations in LB from mCRC patients and that ctDNA may indicate the disappearance of treatment responsive KRAS positive mCRC clones, thereby serving as an early sign of disease progression |
| Ning et al., 2018 [63] | mCRC patients having received standard Food and Drug Administration (FDA)-approved therapies (including fluoropyrimidines, oxaliplatin, irinotecan, bevacizumab, cetuximab, panitumumab, regorafenib), or received experimental agents being tested in three phase I or II clinical trials examining 5-FU plus brivanib (NCT01046864), PRI-724 (NCT01302405) and celecoxib plus EpO906 (NCT00159484). | Measure mRNA expression of EMT (PI3Ka, Akt-2, Twist1) and stem cell (ALDH1) markers in CTCs | -CTCs enrichment: negative immunomagnetic selection using anti- CD45 specific antibodies (Dynabeads M-450 CD45 pan Leukocyte, Invitrogen, Waltham, MA, USA) and then, CD45-negative (CD45−) supernatant s transferred for immune separation and selection using Dynabeads (Dynabeads Epithelial Enrich, #161.02, Invitrogen) with a monoclonal antibody towards human EpCAM,-mRNA expression of EMT (PI3Ka, Akt-2, Twist1) and stem cell (ALDH1) markers was measured | Patients with positive CTC Akt-2 expression had a significantly shorter median PFS in multivariable analyses (HR = 1.70; adjusted p = 0.041) |
| NCT02624726 [64] | Patients treated with FOLFIRI (Folinic acid, fluorouracil and irinotecan)/aflibercept (vascular endothelial growth factor (VEGF) inhibitor) | Objective Response Rate (ORR) | Detection of CEACAM5 mRNA-positive CTCs performed using a real-time PCR assay | At preplanned interim analysis, all patients had discontinued treatment and the ORR was 61.3%, crossing the activity threshold for trial discontinuation.Retaining CTC-negative status predicted better OS compared to continuous detection of CTCs (p = 0.015) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christou, N.; Veyrune, L.; Popeskou, S.G.; Mathonnet, M. Personalized Therapy and Liquid Biopsy—A Focus on Colorectal Cancer. J. Pers. Med. 2021, 11, 630. https://doi.org/10.3390/jpm11070630
Christou N, Veyrune L, Popeskou SG, Mathonnet M. Personalized Therapy and Liquid Biopsy—A Focus on Colorectal Cancer. Journal of Personalized Medicine. 2021; 11(7):630. https://doi.org/10.3390/jpm11070630
Chicago/Turabian StyleChristou, Niki, Léa Veyrune, Sotirios Georgios Popeskou, and Muriel Mathonnet. 2021. "Personalized Therapy and Liquid Biopsy—A Focus on Colorectal Cancer" Journal of Personalized Medicine 11, no. 7: 630. https://doi.org/10.3390/jpm11070630
APA StyleChristou, N., Veyrune, L., Popeskou, S. G., & Mathonnet, M. (2021). Personalized Therapy and Liquid Biopsy—A Focus on Colorectal Cancer. Journal of Personalized Medicine, 11(7), 630. https://doi.org/10.3390/jpm11070630