
Systems Approaches to Treatment Response to Imatinib in Severe Asthma: A Pilot Study

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
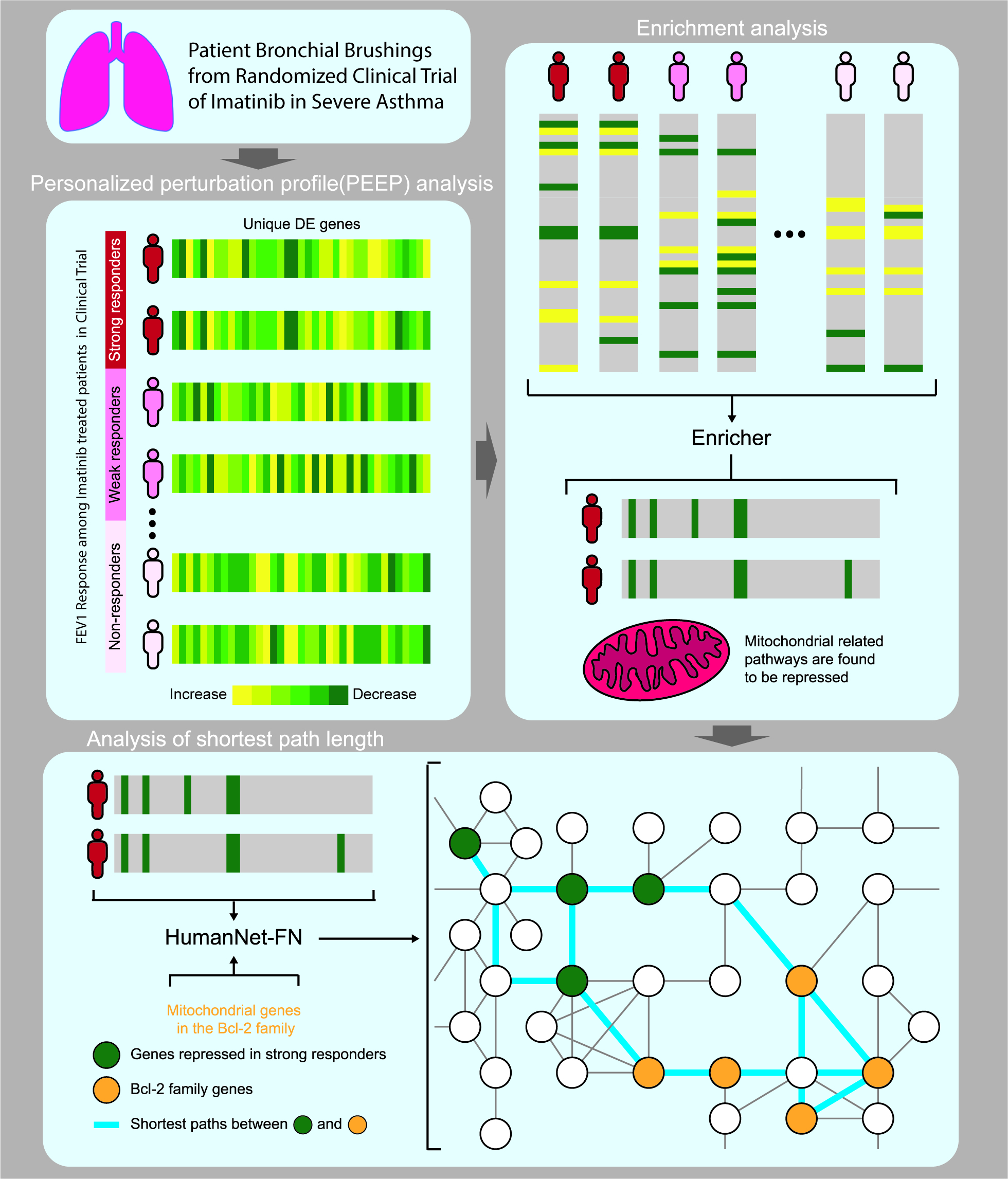
3. Results
3.1. Personalized Perturbation Profile (PEEP) Analysis
3.2. Hierarchical Clustering Based on PEEP
3.3. Shortest Distance Analysis to Mitochondrial Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Carr, T.F.; Kraft, M. Management of Severe Asthma before Referral to the Severe Asthma Specialist. J. Allergy Clin. Immunol. Pr. 2017, 5, 877–886. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. AsthmaStats: Uncontrolled Asthma Among Adults. 2019. Available online: https://www.cdc.gov/asthma/asthma_stats/default.htm (accessed on 7 April 2020).
- Colas, L.; Hassoun, D.; Magnan, A. Needs for Systems Approaches to Better Treat Individuals with Severe Asthma: Predicting Phenotypes and Responses to Treatments. Front. Med. 2020, 7, 7. [Google Scholar] [CrossRef]
- Cahill, K.N.; Katz, H.R.; Cui, J.; Lai, J.; Kazani, S.; Crosby-Thompson, A.; Garofalo, D.; Castro, M.; Jarjour, N.; DiMango, E.; et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N. Engl. J. Med. 2017, 376, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Makowska, J.S.; Cieslak, M.; Kowalski, M.L. Stem cell factor and its soluble receptor (c-kit) in serum of asthmatic patients- correlation with disease severity. BMC Pulm. Med. 2009, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S. Severe Asthma in Adults. Am. J. Respir. Crit. Care Med. 2005, 172, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Baos, S.; Calzada, D.; Cremades-Jimeno, L.; Sastre, J.; Picado, C.; Quiralte, J.; Florido, F.; Lahoz, C.; Cardaba, B. Nonallergic Asthma and Its Severity: Biomarkers for Its Discrimination in Peripheral Samples. Front. Immunol. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.R.; Weiss, S.T.; Glass, K.; Sharma, A. Network Medicine in the Age of Biomedical Big Data. Front. Genet. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Menche, J.; Guney, E.; Sharma, A.; Branigan, P.J.; Loza, M.J.; Baribaud, F.; Dobrin, R.; Barabási, A.-L. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst. Biol. Appl. 2017, 3, 10. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, C.Y.; Yang, S.; Kim, E.; Hart, T.; Marcotte, E.M.; Lee, I. HumanNet v2: Human gene networks for disease research. Nucleic Acids Res. 2019, 47, D573–D580. [Google Scholar] [CrossRef]
- Sharan, R.; Ulitsky, I.; Shamir, R. Network-based prediction of protein function. Mol. Syst. Biol. 2007, 3, 88. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.S.; Wise, R.S.; Covar, R.; Irvin, C.G.; Kercsmar, C.M.; Kraft, M.; Liu, M.C.; O’Connor, G.T.; Peters, S.P.; Sorkness, R.; et al. Asthma outcomes: Pulmonary physiology. J. Allergy Clin. Immunol. 2012, 129, S65–S87. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef]
- Agrawal, A.; Mabaliraian, U. Rejuvenating cellular respiration for optimizing respiratory function: Targeting mitochondria. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L103–L113. [Google Scholar] [CrossRef]
- Delmotte, P.; Sieck, G.C. Interaction between endplasmic/sarcoplasmic reticulum stress (ER/SR stress), mitochondrial signal-ing and Ca(2b) regulation in airway smooth muscle (ASM). Can. J. Physiol. Pharmacol. 2015, 93, 97–110. [Google Scholar] [CrossRef]
- Trian, T.; Benard, G.; Begueret, H.; Rossignol, R.; Girodet, P.-O.; Ghosh, D.; Ousova, O.; Vernejoux, J.-M.; Marthan, R.; Tunon-de-Lara, J.-M.; et al. Bronchial smooth muscle remodeling involves calcium-dependent en-hanced mitochondrial biogenesis in asthma. J. Exp. Med. 2007, 204, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.K.; Bajbouj, K.; Hachim, M.Y.; Mogas, A.K.; Mahboub, B.; Olivenstein, R.; Hamoudi, R.; Halwani, R.; Hamid, Q. Enhanced mitophagy in bronchial fibroblasts from severe asthmatic patients. PLoS ONE 2020, 15, e0242695. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Pinkerton, J.W.; Gibson, P.G.; Cooper, M.A.; Horvat, J.C.; Hansbro, P.M. Inflammasomes in COPD and neutrophilic asthma. Thorax 2015, 70, 1199–1201. [Google Scholar] [CrossRef]
- Cocco, M.P.; White, E.; Xiao, S.; Hu, D.; Mak, A.; Sleiman, P.; Yang, M.; Bobbitt, K.R.; Gui, H.; Levin, A.M.; et al. Asthma and its relationship to mitochondrial copy number: Results from the Asthma Translational Genomics Collaborative (ATGC) of the Trans-Omics for Precision Medicine (TOPMed) program. PLoS ONE 2020, 15, e0242364. [Google Scholar] [CrossRef]
- Xu, W.; Ghosh, S.; Comhair, S.A.; Asosingh, K.; Janocha, A.J.; Mavrakis, D.A.; Bennett, C.D.; Gruca, L.L.; Graham, B.B.; Queisser, K.A.; et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J. Clin. Investig. 2016, 126, 2465–2481. [Google Scholar] [CrossRef] [PubMed]
- Rooswinkel, R.W.; van de Kooij, B.; de Vries, E.; Paauwe, M.; Braster, R.; Verheij, M.; Borst, J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014, 123, 2806–2815. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Hu, Y.; Liu, Y.; Wang, Z.; Geng, H.; Hu, L.; Xu, D.; Wang, K.; Zheng, L.; Zheng, S.; et al. BEX1 Promotes Imatinib-Induced Apoptosis by Binding to and Antagonizing BCL-2. PLoS ONE 2014, 9, e91782. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Denlinger, L.C.; Fowler, S.J.; Akuthota, P.; Shaw, D.E.; Heaney, L.G.; Brown, L.; Castro, M.; Winders, T.A.; Kraft, M.; et al. Unmet Needs in Severe Asthma Subtyping and Precision Medicine Trials. Bridging Clinical and Patient Perspectives. Am. J. Respir. Crit. Care Med. 2019, 199, 823–829. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Imatinib (N = 10) | Placebo (N = 14) |
|---|---|---|
| Age (years) | 44 ± 7 | 41.7 ± 12.3 |
| Female sex (no. %) | 60% | 50% |
| White race ^ (no. %) | 50% | 35.7% |
| Log2 methacholine PC20 ‡ | 1.29 ± 1.5 | 1.47 ± 1.3 |
| FEV1—Percent of predicted | 71.5 ± 12.4 | 68.3 ± 11.7 |
| FeNO ‡—ppb | 23.4 ± 11.5 | 32.4 ± 11 |
| Peripheral eosinophil count, per cubic millimeter | 6.6 ± 7.2 | 2.95 ± 7.2 |
| BAL neutrophils # | 4.2 ± 8.8 | 2.3 ± 5.6 |
| BAL eosinophils & | 0.79 ± 0.81 | 0.54 ± 0.88 |
| ACQ-6 score † | 1.8 ± 1.1 | 2.2 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, S.H.; Foer, D.; Cahill, K.N.; Israel, E.; Maiorino, E.; Röhl, A.; Boyce, J.A.; Weiss, S.T. Systems Approaches to Treatment Response to Imatinib in Severe Asthma: A Pilot Study. J. Pers. Med. 2021, 11, 240. https://doi.org/10.3390/jpm11040240
Baek SH, Foer D, Cahill KN, Israel E, Maiorino E, Röhl A, Boyce JA, Weiss ST. Systems Approaches to Treatment Response to Imatinib in Severe Asthma: A Pilot Study. Journal of Personalized Medicine. 2021; 11(4):240. https://doi.org/10.3390/jpm11040240
Chicago/Turabian StyleBaek, Seung Han, Dinah Foer, Katherine N. Cahill, Elliot Israel, Enrico Maiorino, Annika Röhl, Joshua A. Boyce, and Scott T. Weiss. 2021. "Systems Approaches to Treatment Response to Imatinib in Severe Asthma: A Pilot Study" Journal of Personalized Medicine 11, no. 4: 240. https://doi.org/10.3390/jpm11040240
APA StyleBaek, S. H., Foer, D., Cahill, K. N., Israel, E., Maiorino, E., Röhl, A., Boyce, J. A., & Weiss, S. T. (2021). Systems Approaches to Treatment Response to Imatinib in Severe Asthma: A Pilot Study. Journal of Personalized Medicine, 11(4), 240. https://doi.org/10.3390/jpm11040240