
The Endothelium as a Target for Anti-Atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1



Abstract
1. Introduction
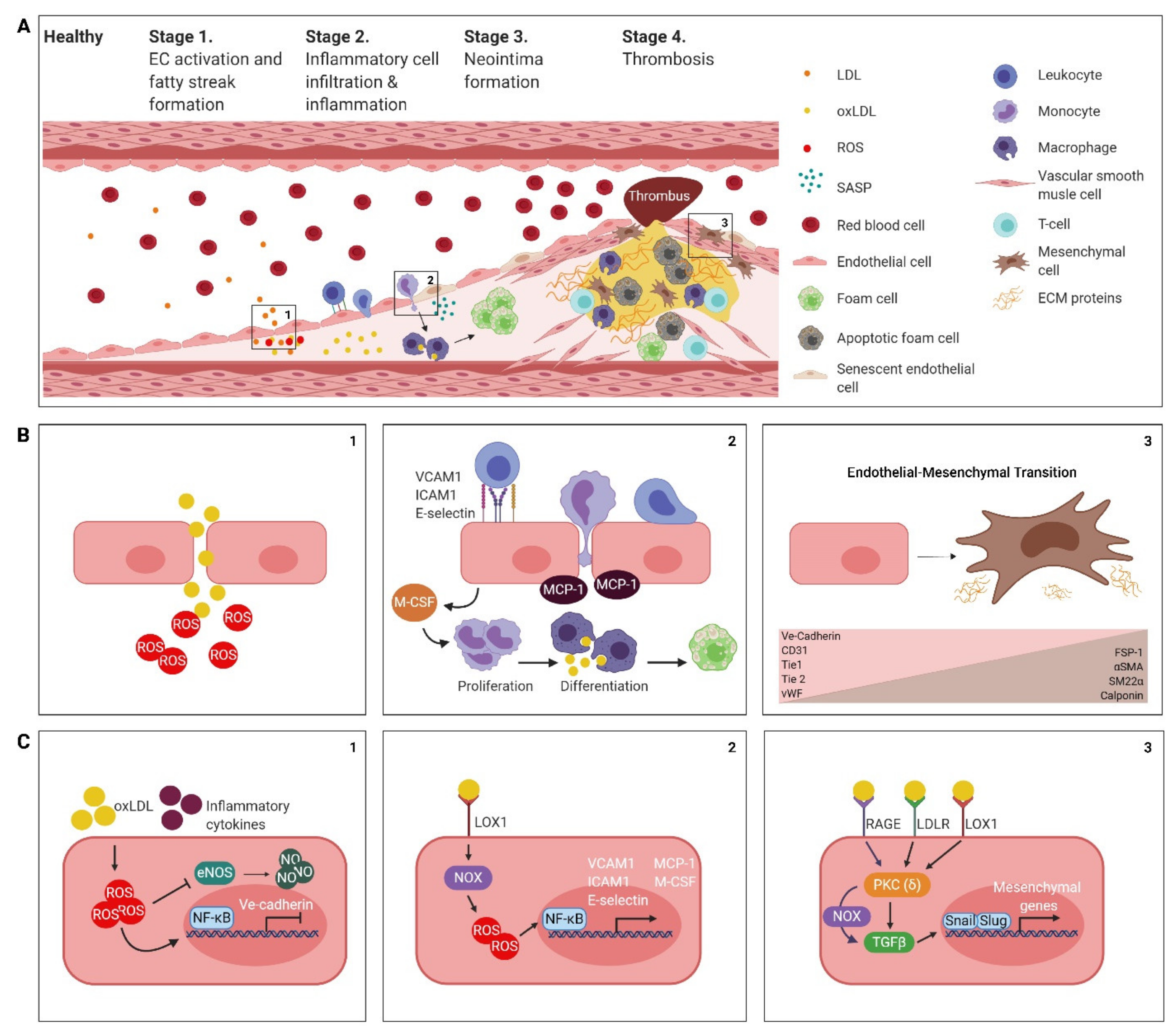
2. The Atherosclerotic Endothelium
2.1. Endothelial Cell Activation, Dysfunction and the Formation of a Fatty Streak
2.2. Inflammatory Cell Infiltration and Inflammation
2.3. Neointima Formation
2.4. Thrombogenesis
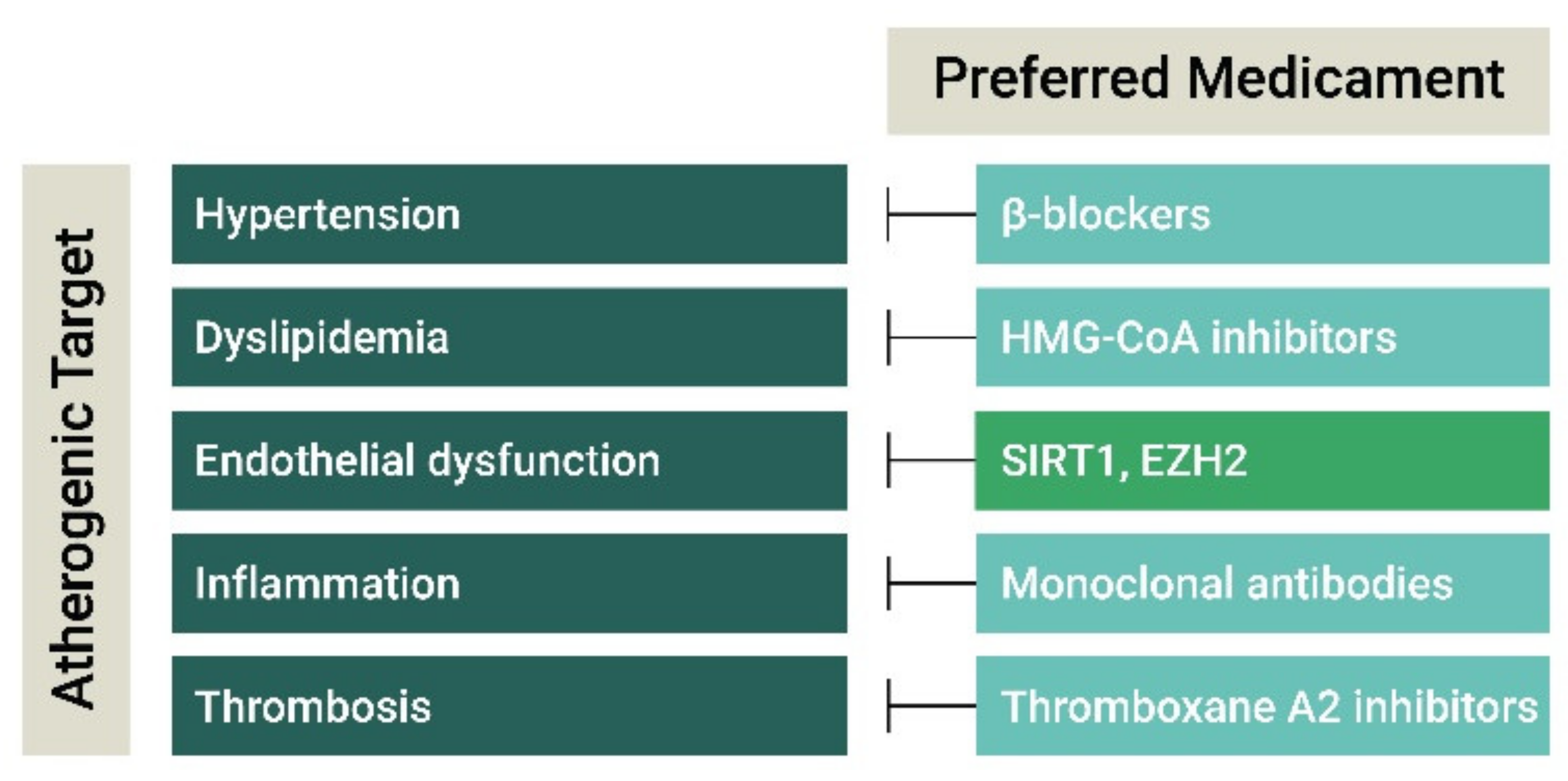
3. Current and Experimental Atherosclerosis Therapies
3.1. Antihypertensive and Lipid-Lowering Drugs
3.2. Anti-Inflammatory Agents
3.3. Anti-Thrombotic Agents
4. The Endothelial Transcriptome as a Target for Anti-Atherogenic Therapy
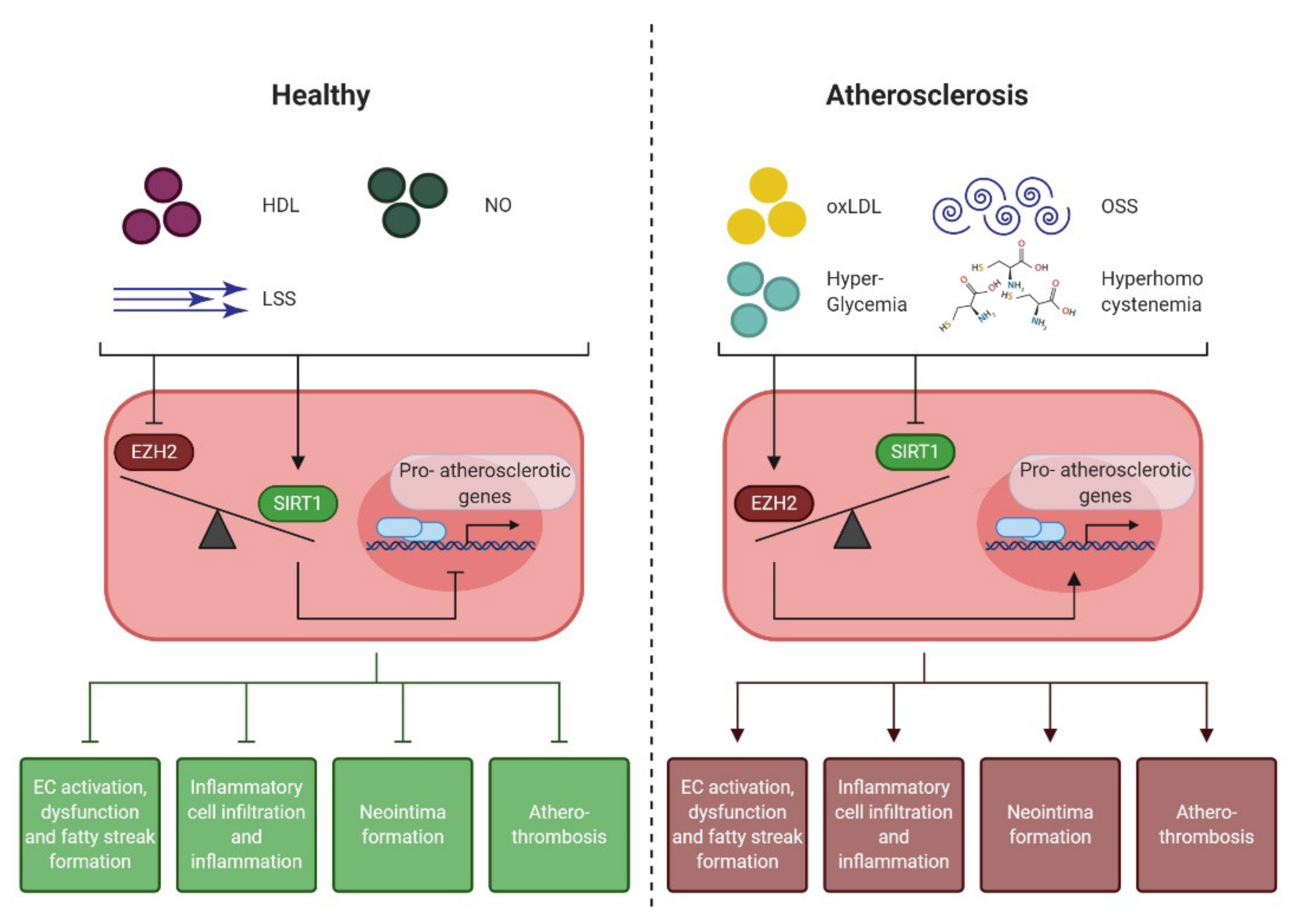
4.1. Epigenetic Regulation of the Endothelial Pro-Atherogenic Phenotype
4.2. Endothelial Enhancer of Zeste Homologue 2 (EZH2)
4.3. Endothelial NAD+-Dependent Deacetylase Sirtuin 1 (SIRT1)
4.4. EZH2 and SIRT1: the Yin and Yang of Early Atherogenesis
5. Future Clinical Perspective
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soliman, G.A. Dietary Fiber, Atherosclerosis, and Cardiovascular Disease. Nutrients 2019, 11, 1155. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Lutgens, E.; van Suylen, R.J.; Faber, B.C.; Gijbels, M.J.; Eurlings, P.M.; Bijnens, A.P.; Cleutjens, K.B.; Heeneman, S.; Daemen, M.J. Atherosclerotic plaque rupture: Local or systemic process? Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2123–2130. [Google Scholar] [CrossRef]
- Ahmadi, A.; Argulian, E.; Leipsic, J.; Newby, D.E.; Narula, J. From Subclinical Atherosclerosis to Plaque Progression and Acute Coronary Events: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1608–1617. [Google Scholar] [CrossRef]
- Gutstein, D.E.; Fuster, V. Pathophysiology and clinical significance of atherosclerotic plaque rupture. Cardiovasc. Res. 1999, 41, 323–333. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr. Vascular endothelium: An integrator of pathophysiologic stimuli in atherosclerosis. Am. J. Cardiol. 1995, 75, 67B–70B. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. The role of ERK5 in endothelial cell function. Biochem. Soc. Trans. 2014, 42, 1584–1589. [Google Scholar] [CrossRef]
- Le, N.T.; Heo, K.S.; Takei, Y.; Lee, H.; Woo, C.H.; Chang, E.; McClain, C.; Hurley, C.; Wang, X.; Li, F.; et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 2013, 127, 486–499. [Google Scholar] [CrossRef]
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef]
- Liu, M.; Chen, X.; Ma, J.; Hassan, W.; Wu, H.; Ling, J.; Shang, J. beta-Elemene attenuates atherosclerosis in apolipoprotein E-deficient mice via restoring NO levels and alleviating oxidative stress. Biomed. Pharmacother. 2017, 95, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Fang, G.; Mao, S.Z.; Ye, X.; Liu, G.; Miller, E.J.; Greenberg, H.; Liu, S.F. Selective inhibition of endothelial NF-kappaB signaling attenuates chronic intermittent hypoxia-induced atherosclerosis in mice. Atherosclerosis 2018, 270, 68–75. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Smith, S.C., Jr.; Stone, N.J.; Grundy, S.M. Secondary Prevention for Atherosclerotic Cardiovascular Disease: Comparing Recent US and European Guidelines on Dyslipidemia. Circulation 2020, 141, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Al Suwaidi, J.; Hamasaki, S.; Higano, S.T.; Nishimura, R.A.; Holmes, D.R.; Lerman, A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000, 101, 948–954. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Boullier, A.; Bird, D.A.; Chang, M.K.; Dennis, E.A.; Friedman, P.; Gillotre-Taylor, K.; Horkko, S.; Palinski, W.; Quehenberger, O.; Shaw, P.; et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 214–222. [Google Scholar] [CrossRef]
- Itabe, H. Oxidative modification of LDL: Its pathological role in atherosclerosis. Clin. Rev. Allergy Immunol. 2009, 37, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Corson, M.A.; James, N.L.; Latta, S.E.; Nerem, R.M.; Berk, B.C.; Harrison, D.G. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ. Res. 1996, 79, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Oemar, B.S.; Tschudi, M.R.; Godoy, N.; Brovkovich, V.; Malinski, T.; Luscher, T.F. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation 1998, 97, 2494–2498. [Google Scholar] [CrossRef]
- Ryoo, S.; Lemmon, C.A.; Soucy, K.G.; Gupta, G.; White, A.R.; Nyhan, D.; Shoukas, A.; Romer, L.H.; Berkowitz, D.E. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ. Res. 2006, 99, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Takei, A.; Huang, Y.; Lopes-Virella, M.F. Expression of adhesion molecules by human endothelial cells exposed to oxidized low density lipoprotein. Influences of degree of oxidation and location of oxidized LDL. Atherosclerosis 2001, 154, 79–86. [Google Scholar] [CrossRef]
- Amberger, A.; Maczek, C.; Jurgens, G.; Michaelis, D.; Schett, G.; Trieb, K.; Eberl, T.; Jindal, S.; Xu, Q.; Wick, G. Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous endothelial cells in response to cytokines and oxidized low-density lipoproteins. Cell Stress Chaperones 1997, 2, 94–103. [Google Scholar] [CrossRef]
- Shi, W.; Haberland, M.E.; Jien, M.L.; Shih, D.M.; Lusis, A.J. Endothelial responses to oxidized lipoproteins determine genetic susceptibility to atherosclerosis in mice. Circulation 2000, 102, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Dansky, H.M.; Barlow, C.B.; Lominska, C.; Sikes, J.L.; Kao, C.; Weinsaft, J.; Cybulsky, M.I.; Smith, J.D. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1662–1667. [Google Scholar] [CrossRef]
- Kleemann, R.; Zadelaar, S.; Kooistra, T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc. Res. 2008, 79, 360–376. [Google Scholar] [CrossRef]
- Bobryshev, Y.V. Monocyte recruitment and foam cell formation in atherosclerosis. Micron 2006, 37, 208–222. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Stojanovic, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Scott-Burden, T.; Vanhoutte, P.M. Regulation of smooth muscle cell growth by endothelium-derived factors. Tex. Heart Inst. J. 1994, 21, 91–97. [Google Scholar]
- Krenning, G.; Barauna, V.G.; Krieger, J.E.; Harmsen, M.C.; Moonen, J.R. Endothelial Plasticity: Shifting Phenotypes through Force Feedback. Stem Cells Int. 2016, 2016, 9762959. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Wesseling, M.; Sakkers, T.R.; de Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vasc. Pharmacol. 2018, 106, 1–8. [Google Scholar] [CrossRef]
- Mahmoud, M.M.; Kim, H.R.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Serbanovic-Canic, J.; Feng, S.; Bowden, N.P.; Maguire, R.; et al. TWIST1 Integrates Endothelial Responses to Flow in Vascular Dysfunction and Atherosclerosis. Circ. Res. 2016, 119, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff, S. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2278–2289. [Google Scholar] [CrossRef]
- Clarke, M.C.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Shah, P.K.; Falk, E.; Badimon, J.J.; Fernandez-Ortiz, A.; Mailhac, A.; Villareal-Levy, G.; Fallon, J.T.; Regnstrom, J.; Fuster, V. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 1995, 92, 1565–1569. [Google Scholar] [PubMed]
- Vendrov, A.E.; Stevenson, M.D.; Alahari, S.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. Attenuated Superoxide Dismutase 2 Activity Induces Atherosclerotic Plaque Instability During Aging in Hyperlipidemic Mice. J. Am. Heart Assoc. 2017, 6, e006775. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, Z.; Dong, Z.; Wang, C.; Cao, Q.; Fan, F.; Zhao, J.; Liu, X.; Yuan, M.; Sun, X.; et al. Aldehyde dehydrogenase 2 deficiency promotes atherosclerotic plaque instability through accelerating mitochondrial ROS-mediated vascular smooth muscle cell senescence. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1782–1792. [Google Scholar] [CrossRef]
- Gordon, D.; Reidy, M.A.; Benditt, E.P.; Schwartz, S.M. Cell proliferation in human coronary arteries. Proc. Natl. Acad. Sci. USA 1990, 87, 4600–4604. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.R.; Alpers, C.E.; Stewart, D.K.; Ferguson, M.; Tran, N.; Gordon, D.; Benditt, E.P.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M. Proliferation in primary and restenotic coronary atherectomy tissue. Implications for antiproliferative therapy. Circ. Res. 1993, 73, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Susic, D. Hypertension, aging, and atherosclerosis. The endothelial interface. Med. Clin. N. Am. 1997, 81, 1231–1240. [Google Scholar] [CrossRef]
- James, P.A.; Oparil, S.; Carter, B.L.; Cushman, W.C.; Dennison-Himmelfarb, C.; Handler, J.; Lackland, D.T.; LeFevre, M.L.; MacKenzie, T.D.; Ogedegbe, O.; et al. 2014 evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014, 311, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Olafiranye, O.; Zizi, F.; Brimah, P.; Jean-Louis, G.; Makaryus, A.N.; McFarlane, S.; Ogedegbe, G. Management of Hypertension among Patients with Coronary Heart Disease. Int. J. Hypertens. 2011, 2011, 653903. [Google Scholar] [CrossRef]
- Riccioni, G.; Sblendorio, V. Atherosclerosis: From biology to pharmacological treatment. J. Geriatr. Cardiol. JGC 2012, 9, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S. Antihypertensive drugs. Pharmacol. Res. 2017, 124, 116–125. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Dézsi, C.A. The Different Therapeutic Choices with ARBs. Which One to Give? When? Why? Am. J. Cardiovasc. Drugs 2016, 16, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIb randomized placebo controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Schmitt, C.; Abt, M.; Ciorciaro, C.; Kling, D.; Jamois, C.; Schick, E.; Solier, C.; Benghozi, R.; Gaudreault, J. First-in-Man Study With Inclacumab, a Human Monoclonal Antibody Against P-selectin. J. Cardiovasc. Pharmacol. 2015, 65, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Ibrahim, R.; Gregoire, J.C.; L’Allier, P.L.; Pressacco, J.; Tardif, J.C.; Budoff, M.J. Effect of treatment with 5-lipoxygenase inhibitor VIA-2291 (atreleuton) on coronary plaque progression: A serial CT angiography study. Clin. Cardiol. 2017, 40, 210–215. [Google Scholar] [CrossRef]
- Back, M. Inhibitors of the 5-lipoxygenase pathway in atherosclerosis. Curr. Pharm. Des. 2009, 15, 3116–3132. [Google Scholar] [CrossRef]
- Serruys, P.W.; Garcia-Garcia, H.M.; Buszman, P.; Erne, P.; Verheye, S.; Aschermann, M.; Duckers, H.; Bleie, O.; Dudek, D.; Botker, H.E.; et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation 2008, 118, 1172–1182. [Google Scholar] [CrossRef]
- Leite, J.O.; Vaishnav, U.; Puglisi, M.; Fraser, H.; Trias, J.; Fernandez, M.L. A-002 (Varespladib), a phospholipase A2 inhibitor, reduces atherosclerosis in guinea pigs. BMC Cardiovasc. Disord. 2009, 9, 7. [Google Scholar] [CrossRef]
- Winter, C.; Silvestre-Roig, C.; Ortega-Gomez, A.; Lemnitzer, P.; Poelman, H.; Schumski, A.; Winter, J.; Drechsler, M.; de Jong, R.; Immler, R.; et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab. 2018, 28, 175–182.e5. [Google Scholar] [CrossRef] [PubMed]
- Gallone, G.; Baldetti, L.; Pagnesi, M.; Latib, A.; Colombo, A.; Libby, P.; Giannini, F. Medical Therapy for Long-Term Prevention of Atherothrombosis Following an Acute Coronary Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2886–2903. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Morais, J.; Baigent, C.; Collet, J.P.; Fitzgerald, D.; Halvorsen, S.; Rocca, B.; Siegbahn, A.; Storey, R.F.; Vilahur, G. Antiplatelet Agents for the Treatment and Prevention of Coronary Atherothrombosis. J. Am. Coll. Cardiol. 2017, 70, 1760–1776. [Google Scholar] [CrossRef] [PubMed]
- European Association for Cardiovascular, Prevention, Rehabilitation; Reiner, Z.; Catapano, A.L.; De Backer, G.; Graham, I.; Taskinen, M.R.; Wiklund, O.; Agewall, S.; Alegria, E.; Chapman, M.J.; et al. ESC/EAS Guidelines for the management of dyslipidaemias: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur. Heart J. 2011, 32, 1769–1818. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur. Heart J. 2016, 37, 1720–1722. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Clearfield, M.; Downs, J.R.; Weis, S.E.; Miles, J.S.; Gotto, A.M., Jr.; Air Force/Texas Coronary Atherosclerosis Prevention Study Investigators. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Engl. J. Med. 2001, 344, 1959–1965. [Google Scholar] [CrossRef]
- Dadu, R.T.; Ballantyne, C.M. Lipid lowering with PCSK9 inhibitors. Nat. Rev. Cardiol. 2014, 11, 563–575. [Google Scholar] [CrossRef]
- Garcia-Calvo, M.; Lisnock, J.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.W.; Ramji, D.P. Nutraceutical therapies for atherosclerosis. Nat. Rev. Cardiol. 2016, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 281–290. [Google Scholar] [CrossRef]
- Zhang, X.; Niessner, A.; Nakajima, T.; Ma-Krupa, W.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. Interleukin 12 induces T-cell recruitment into the atherosclerotic plaque. Circ. Res. 2006, 98, 524–531. [Google Scholar] [CrossRef]
- Halvorsen, B.; Waehre, T.; Scholz, H.; Clausen, O.P.; von der Thusen, J.H.; Muller, F.; Heimli, H.; Tonstad, S.; Hall, C.; Froland, S.S.; et al. Interleukin-10 enhances the oxidized LDL-induced foam cell formation of macrophages by antiapoptotic mechanisms. J. Lipid Res. 2005, 46, 211–219. [Google Scholar] [CrossRef]
- Fu, H.; Tang, Y.Y.; Ouyang, X.P.; Tang, S.L.; Su, H.; Li, X.; Huang, L.P.; He, M.; Lv, Y.C.; He, P.P.; et al. Interleukin-27 inhibits foam cell formation by promoting macrophage ABCA1 expression through JAK2/STAT3 pathway. Biochem. Biophys. Res. Commun. 2014, 452, 881–887. [Google Scholar] [CrossRef]
- Sanada, F.; Muratsu, J.; Otsu, R.; Shimizu, H.; Koibuchi, N.; Uchida, K.; Taniyama, Y.; Yoshimura, S.; Rakugi, H.; Morishita, R. Local Production of Activated Factor X in Atherosclerotic Plaque Induced Vascular Smooth Muscle Cell Senescence. Sci. Rep. 2017, 7, 17172. [Google Scholar] [CrossRef]
- Jain, M.; Singh, A.; Singh, V.; Barthwal, M.K. Involvement of interleukin-1 receptor-associated kinase-1 in vascular smooth muscle cell proliferation and neointimal formation after rat carotid injury. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1445–1455. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H282–H296. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Taub, R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat. Rev. Drug Discov. 2011, 10, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Grechko, A.V.; Myasoedova, V.A.; Orekhov, A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp. Mol. Pathol. 2018, 104, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Meadows, T.A.; Bhatt, D.L. Clinical aspects of platelet inhibitors and thrombus formation. Circ. Res. 2007, 100, 1261–1275. [Google Scholar] [CrossRef]
- Weksler, B.B.; Marcus, A.J.; Jaffe, E.A. Synthesis of prostaglandin I2 (prostacyclin) by cultured human and bovine endothelial cells. Proc. Natl. Acad. Sci. USA 1977, 74, 3922–3926. [Google Scholar] [CrossRef]
- Chan, V.; Chan, T.K. Antithrombin III in fresh and cultured human endothelial cells: A natural anticoagulant from the vascular endothelium. Thromb. Res. 1979, 15, 209–213. [Google Scholar] [CrossRef]
- Wei, H.J.; Li, Y.H.; Shi, G.Y.; Liu, S.L.; Chang, P.C.; Kuo, C.H.; Wu, H.L. Thrombomodulin domains attenuate atherosclerosis by inhibiting thrombin-induced endothelial cell activation. Cardiovasc. Res. 2011, 92, 317–327. [Google Scholar] [CrossRef]
- Loscalzo, J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ. Res. 2001, 88, 756–762. [Google Scholar] [CrossRef]
- Goldstein, L.B.; Adams, R.; Alberts, M.J.; Appel, L.J.; Brass, L.M.; Bushnell, C.D.; Culebras, A.; DeGraba, T.J.; Gorelick, P.B.; Guyton, J.R.; et al. Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2006, 113, e873–e923. [Google Scholar] [CrossRef]
- Erbilgin, A.; Siemers, N.; Kayne, P.; Yang, W.P.; Berliner, J.; Lusis, A.J. Gene expression analyses of mouse aortic endothelium in response to atherogenic stimuli. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2509–2517. [Google Scholar] [CrossRef]
- Deng, D.X.; Tsalenko, A.; Vailaya, A.; Ben-Dor, A.; Kundu, R.; Estay, I.; Tabibiazar, R.; Kincaid, R.; Yakhini, Z.; Bruhn, L.; et al. Differences in vascular bed disease susceptibility reflect differences in gene expression response to atherogenic stimuli. Circ. Res. 2006, 98, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Andalibi, A.; deBeer, F.C.; Fogelman, A.M.; Lusis, A.J. Genetic control of inflammatory gene induction and NF-kappa B-like transcription factor activation in response to an atherogenic diet in mice. J. Clin. Investig. 1993, 91, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Civelek, M.; Fang, Y.; Fleming, I. The atherosusceptible endothelium: Endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 2013, 99, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Greissel, A.; Culmes, M.; Burgkart, R.; Zimmermann, A.; Eckstein, H.H.; Zernecke, A.; Pelisek, J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2016, 25, 79–86. [Google Scholar] [CrossRef]
- Xu, S.; Xu, Y.; Yin, M.; Zhang, S.; Liu, P.; Koroleva, M.; Si, S.; Little, P.J.; Pelisek, J.; Jin, Z.G. Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects. Theranostics 2018, 8, 3007–3021. [Google Scholar] [CrossRef]
- Lv, Y.C.; Tang, Y.Y.; Zhang, P.; Wan, W.; Yao, F.; He, P.P.; Xie, W.; Mo, Z.C.; Shi, J.F.; Wu, J.F.; et al. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PLoS ONE 2016, 11, e0157265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Virdis, A.; Hussain, S.; Mohammed, S.A.; Capretti, G.; Akhmedov, A.; Dalgaard, K.; Chiandotto, S.; Pospisilik, J.A.; et al. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur. Heart J. 2019, 40, 383–391. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef]
- Dreger, H.; Ludwig, A.; Weller, A.; Stangl, V.; Baumann, G.; Meiners, S.; Stangl, K. Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension 2012, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Maleszewska, M.; Vanchin, B.; Harmsen, M.C.; Krenning, G. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis 2016, 19, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Vikram, A.; Hoffman, T.A.; Naqvi, A.; Lewarchik, C.M.; Kim, Y.R.; Irani, K. Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1936–1942. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Li, C.; Zhang, Y.; Zhou, X.; Lu, C. Long noncoding RNA OIP5-AS1 accelerates the ox-LDL mediated vascular endothelial cells apoptosis through targeting GSK-3beta via recruiting EZH2. Am. J. Transl. Res. 2019, 11, 1827–1834. [Google Scholar]
- Yang, M.; Lv, H.; Liu, Q.; Zhang, L.; Zhang, R.; Huang, X.; Wang, X.; Han, B.; Hou, S.; Liu, D.; et al. Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 9173530. [Google Scholar] [CrossRef]
- Hung, C.H.; Chan, S.H.; Chu, P.M.; Tsai, K.L. Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol. Nutr. Food Res. 2015, 59, 1905–1917. [Google Scholar] [CrossRef]
- Pan, W.; Yu, H.; Huang, S.; Zhu, P. Resveratrol Protects against TNF-alpha-Induced Injury in Human Umbilical Endothelial Cells through Promoting Sirtuin-1-Induced Repression of NF-KB and p38 MAPK. PLoS ONE 2016, 11, e0147034. [Google Scholar] [CrossRef]
- Tsai, K.L.; Hung, C.H.; Chan, S.H.; Hsieh, P.L.; Ou, H.C.; Cheng, Y.H.; Chu, P.M. Chlorogenic Acid Protects Against oxLDL-Induced Oxidative Damage and Mitochondrial Dysfunction by Modulating SIRT1 in Endothelial Cells. Mol. Nutr. Food Res. 2018, 62, e1700928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Sun, X.; Lv, S.; Sun, M.; Guo, H.; Zhai, Y.; Wang, Z.; Dai, P.; Zheng, L.; Ye, M.; et al. Salidroside attenuates oxidized lowdensity lipoproteininduced endothelial cell injury via promotion of the AMPK/SIRT1 pathway. Int. J. Mol. Med. 2019, 43, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Villarreal, G., Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res. 2010, 85, 514–519. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef]
- Luo, Y.; Fang, Y.; Kang, R.; Lenahan, C.; Gamdzyk, M.; Zhang, Z.; Okada, T.; Tang, J.; Chen, S.; Zhang, J.H. Inhibition of EZH2 (Enhancer of Zeste Homolog 2) Attenuates Neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-kappaB (Trimethylation of Histone 3 Lysine 27/Suppressor of Cytokine Signaling 3/Tumor Necrosis Factor Receptor Family 6/Nuclear Factor-kappaB) in a Rat Model of Subarachnoid Hemorrhage. Stroke 2020, 51, 3320–3331. [Google Scholar] [CrossRef]
- Wang, J.; Li, P.; Xu, X.; Zhang, B.; Zhang, J. MicroRNA-200a Inhibits Inflammation and Atherosclerotic Lesion Formation by Disrupting EZH2-Mediated Methylation of STAT3. Front. Immunol. 2020, 11, 907. [Google Scholar] [CrossRef]
- Stein, S.; Schafer, N.; Breitenstein, A.; Besler, C.; Winnik, S.; Lohmann, C.; Heinrich, K.; Brokopp, C.E.; Handschin, C.; Landmesser, U.; et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging 2010, 2, 353–360. [Google Scholar] [CrossRef]
- Chen, H.; Wan, Y.; Zhou, S.; Lu, Y.; Zhang, Z.; Zhang, R.; Chen, F.; Hao, D.; Zhao, X.; Guo, Z.; et al. Endothelium-specific SIRT1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci. China Life Sci. 2012, 55, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Chen, L.K.; Chang, Y.L.; Yung, M.C.; Hsu, C.C.; Chen, Y.C.; Lo, W.L.; Chen, S.J.; Ku, H.H.; Hwang, S.J. Resveratrol protects human endothelium from H(2)O(2)-induced oxidative stress and senescence via SirT1 activation. J. Atheroscler. Thromb. 2010, 17, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Wu, H.; Wu, J.; Zhou, S.; Huang, W.; Li, Y.; Zhang, H.; Wang, J.; Jia, Y. SRT2104 attenuates diabetes-induced aortic endothelial dysfunction via inhibition of P53. J. Endocrinol. 2018, 237, 1–14. [Google Scholar] [CrossRef]
- Yang, R.; Fang, W.; Liang, J.; Lin, C.; Wu, S.; Yan, S.; Hu, C.; Ke, X. Apelin/APJ axis improves angiotensin II-induced endothelial cell senescence through AMPK/SIRT1 signaling pathway. Arch. Med. Sci. AMS 2018, 14, 725–734. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Zha, S.; Cao, Q.; Sheng, J.; Chen, S. SIRT1 inhibits TGF-beta-induced endothelial-mesenchymal transition in human endothelial cells with Smad4 deacetylation. J. Cell. Physiol. 2018, 233, 9007–9014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Zhang, Y.; Wang, X.; Fan, X.F.; Zhang, Y.; Li, X.; Gong, Y.S.; Han, L.P. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed. Pharmacother. 2019, 118, 109227. [Google Scholar] [CrossRef]
- Lipphardt, M.; Dihazi, H.; Muller, G.A.; Goligorsky, M.S. Fibrogenic Secretome of Sirtuin 1-Deficient Endothelial Cells: Wnt, Notch and Glycocalyx Rheostat. Front. Physiol. 2018, 9, 1325. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1634–1639. [Google Scholar] [CrossRef]
- Wan, Y.Z.; Gao, P.; Zhou, S.; Zhang, Z.Q.; Hao, D.L.; Lian, L.S.; Li, Y.J.; Chen, H.Z.; Liu, D.P. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 2014, 13, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, M.C.; Liang, M.; Fu, J. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood 2012, 119, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Piazzolla, G.; Candigliota, M.; Fanelli, M.; Castrovilli, A.; Berardi, E.; Antonica, G.; Battaglia, S.; Solfrizzi, V.; Sabba, C.; Tortorella, C. Hyperhomocysteinemia is an independent risk factor of atherosclerosis in patients with metabolic syndrome. Diabetol. Metab. Syndr. 2019, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Xiaoling, Y.; Li, Z.; ShuQiang, L.; Shengchao, M.; Anning, Y.; Ning, D.; Nan, L.; Yuexia, J.; Xiaoming, Y.; Guizhong, L. Hyperhomocysteinemia in ApoE-/-mice leads to overexpression of enhancer of Zeste Homolog 2 via miR-92a regulation. PLoS ONE 2016, 11, e0167744. [Google Scholar] [CrossRef] [PubMed]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef]
- Vignini, A.; Nanetti, L.; Bacchetti, T.; Ferretti, G.; Curatola, G.; Mazzanti, L. Modification induced by homocysteine and low-density lipoprotein on human aortic endothelial cells: An in vitro study. J. Clin. Endocrinol. Metab. 2004, 89, 4558–4561. [Google Scholar] [CrossRef]
- Brooks, A.R.; Lelkes, P.I.; Rubanyi, G.M. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol. Genom. 2002, 9, 27–41. [Google Scholar] [CrossRef]
- Passerini, A.G.; Polacek, D.C.; Shi, C.; Francesco, N.M.; Manduchi, E.; Grant, G.R.; Pritchard, W.F.; Powell, S.; Chang, G.Y.; Stoeckert, C.J., Jr.; et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc. Natl. Acad. Sci. USA 2004, 101, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Boldo, L.S.; Fernandez, B.O.; Feelisch, M.; Harmsen, M.C. Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF-β1. Sci. Rep. 2017, 7, 42487. [Google Scholar] [CrossRef] [PubMed]
- Van Thienen, J.V.; Fledderus, J.O.; Dekker, R.J.; Rohlena, J.; van Ijzendoorn, G.A.; Kootstra, N.A.; Pannekoek, H.; Horrevoets, A.J. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc. Res. 2006, 72, 231–240. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef]
- Luo, H.; Jiang, Y.; Ma, S.; Chang, H.; Yi, C.; Cao, H.; Gao, Y.; Guo, H.; Hou, J.; Yan, J.; et al. EZH2 promotes invasion and metastasis of laryngeal squamous cells carcinoma via epithelial-mesenchymal transition through H3K27me3. Biochem. Biophys. Res. Commun. 2016, 479, 253–259. [Google Scholar] [CrossRef]
- Zhao, M.; Hu, X.; Xu, Y.; Wu, C.; Chen, J.; Ren, Y.; Kong, L.; Sun, S.; Zhang, L.; Jin, R.; et al. Targeting of EZH2 inhibits epithelial-mesenchymal transition in head and neck squamous cell carcinoma via regulating the STAT3/VEGFR2 axis. Int. J. Oncol. 2019, 55, 1165–1175. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, Z.; Zhang, X. Effects of miR1955p on cell proliferation and apoptosis in gestational diabetes mellitus via targeting EZH2. Mol. Med. Rep. 2020, 22, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.; Mir, S.E.; Nilsson, R.J.; van der Stoop, P.M.; Niers, J.M.; Marquez, V.E.; Cloos, J.; Breakefield, X.O.; Krichevsky, A.M.; Noske, D.P.; et al. Down-regulation of miR-101 in endothelial cells promotes blood vessel formation through reduced repression of EZH2. PLoS ONE 2011, 6, e16282. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol. Ther. 2016, 165, 26–31. [Google Scholar] [CrossRef]
- Vella, S.; Pomella, S.; Leoncini, P.P.; Colletti, M.; Conti, B.; Marquez, V.E.; Strillacci, A.; Roma, J.; Gallego, S.; Milano, G.M.; et al. MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma. Clin. Epigenetics 2015, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Miranda, M.X.; van Tits, L.J.; Lohmann, C.; Arsiwala, T.; Winnik, S.; Tailleux, A.; Stein, S.; Gomes, A.P.; Suri, V.; Ellis, J.L.; et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur. Heart J. 2015, 36, 51–59. [Google Scholar] [CrossRef]
- Li, J.; Zhong, Z.; Yuan, J.; Chen, X.; Huang, Z.; Wu, Z. Resveratrol improves endothelial dysfunction and attenuates atherogenesis in apolipoprotein E-deficient mice. J. Nutr. Biochem. 2019, 67, 63–71. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Bloch, K.D. The vascular biology of nitric oxide and its role in atherogenesis. Annu. Rev. Med. 1996, 47, 365–375. [Google Scholar] [CrossRef]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar] [CrossRef]
- Meng, X.D.; Yao, H.H.; Wang, L.M.; Yu, M.; Shi, S.; Yuan, Z.X.; Liu, J. Knockdown of GAS5 Inhibits Atherosclerosis Progression via Reducing EZH2-Mediated ABCA1 Transcription in ApoE(-/-) Mice. Mol. Ther. Nucleic Acids 2020, 19, 84–96. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Schafer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Boren, J.; et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur. Heart J. 2010, 31, 2301–2309. [Google Scholar] [CrossRef]
- Wakeling, L.A.; Ions, L.J.; Escolme, S.M.; Cockell, S.J.; Su, T.; Dey, M.; Hampton, E.V.; Jenkins, G.; Wainwright, L.J.; McKay, J.A.; et al. SIRT1 affects DNA methylation of polycomb group protein target genes, a hotspot of the epigenetic shift observed in ageing. Hum. Genom. 2015, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Cano-Rodriguez, D.; Gjaltema, R.A.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Rots, M.G. Epigenetic Editing: On the Verge of Reprogramming Gene Expression at Will. Curr. Genet. Med. Rep. 2016, 4, 170–179. [Google Scholar] [CrossRef]
- Ross, J.S.; Stagliano, N.E.; Donovan, M.J.; Breitbart, R.E.; Ginsburg, G.S. Atherosclerosis and Cancer. Ann. N. Y. Acad. Sci. 2006, 947, 271–293. [Google Scholar] [CrossRef]
- Xu, L.; Wang, R.; Liu, H.; Wang, J.; Mang, J.; Xu, Z. Resveratrol Treatment Is Associated with Lipid Regulation and Inhibition of Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) in Rabbits Fed a High-Fat Diet. Evid. Based Complementary Altern. Med. eCAM 2020, 2020, 9641582. [Google Scholar] [CrossRef]
- Zhou, L.; Long, J.; Sun, Y.; Chen, W.; Qiu, R.; Yuan, D. Resveratrol ameliorates atherosclerosis induced by high-fat diet and LPS in ApoE(-/-) mice and inhibits the activation of CD4(+) T cells. Nutr. Metab. 2020, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Kumar, K.S.; Krishnan, K.; Antony, H. Quercetin alleviates hypercholesterolemic diet induced inflammation during progression and regression of atherosclerosis in rabbits. Nutrition 2013, 29, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Jia, Q.; Shen, D.; Yan, L.; Chen, C.; Xing, S. Quercetin has a protective effect on atherosclerosis via enhancement of autophagy in ApoE(-/-) mice. Exp. Ther. Med. 2019, 18, 2451–2458. [Google Scholar] [CrossRef]
- Kleemann, R.; Verschuren, L.; Morrison, M.; Zadelaar, S.; van Erk, M.J.; Wielinga, P.Y.; Kooistra, T. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis 2011, 218, 44–52. [Google Scholar] [CrossRef]
- Nie, J.; Zhang, L.; Zhao, G.; Du, X. Quercetin reduces atherosclerotic lesions by altering the gut microbiota and reducing atherogenic lipid metabolites. J. Appl. Microbiol. 2019, 127, 1824–1834. [Google Scholar] [CrossRef]
- Chen, Y.X.; Zhang, M.; Cai, Y.; Zhao, Q.; Dai, W. The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE(-)/(-) mice through inhibiting vascular inflammatory response. Biochem. Biophys. Res. Commun. 2015, 465, 732–738. [Google Scholar] [CrossRef]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Joshi, N.V.; Mills, N.L.; Hoffmann, E.; Jacobson, E.W.; Vlasuk, G.P.; Waterhouse, B.R.; et al. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J. Am. Heart Assoc. 2013, 2, e000042. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Boesch-Saadatmandi, C.; Wolffram, S.; Rimbach, G.; Muller, M.J. Serum lipid and blood pressure responses to quercetin vary in overweight patients by apolipoprotein E genotype. J. Nutr. 2010, 140, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Breitenstein, A.; Wyss, C.A.; Spescha, R.D.; Franzeck, F.C.; Hof, D.; Riwanto, M.; Hasun, M.; Akhmedov, A.; von Eckardstein, A.; Maier, W.; et al. Peripheral blood monocyte Sirt1 expression is reduced in patients with coronary artery disease. PLoS ONE 2013, 8, e53106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Atherogenic Target | Class | Compound | Molecular Target | Stage of Development | Refs |
|---|---|---|---|---|---|
| Hypertension | β-blockers | Metoprolol, Carvedilol, Bisoprolol | adrenergic β-receptors | Marketed | [56,57,60,61] |
| ACE inhibitors | Captopril, Benazepril, Perindopril, Ramipril | Angiotensin-converting enzyme | |||
| Ca2+-channel blockers | Amlodipine, Nifedipine | Voltage-dependent L−, N−, and T-type Ca2+ channels | |||
| Diuretics | Thiazide | Solute carrier family 12 members | |||
| Angiotensin-Receptor blockers | Losartan, Valsartan | Angiotensin receptor | Marketed | [60,61,62,63] | |
| Dyslipidemia | HGM-CoA inhibitors | Statins | HMG-CoA | Marketed | [17,62] |
| PCSK9 inhibitors | Evolucumab, Alirocumab | PCSK9 | |||
| Cholesterol absorption inhibitors | Ezetimibe | NPC1L1, SOAT1 | |||
| Inflammation | Antibodies | Canakinumab; Adalimumab, Infliximab, Inclacumab | Cytokines (IL1β, TNFα), adhesion molecules (P-selectin) | I-III | [64,65,66] |
| Lipoxygenase inhibitors | Atreleutron, Veliflapon | 5-LO, FLAP | II | [67,68] | |
| Phospholipase inhibitors | Darapladib, Varespladib | Lp-PLA2, sPLA2 | III | [69,70] | |
| CCL2-CCR2 inhibitors | CCR2 | I | [71] | ||
| Thrombosis | Thromboxane A2 inhibitors | Aspirin | Cyclooxygenases | Marketed | [72,73] |
| P2Y12 inhibitors | Clopidegril, Ticagrelor, Prasugrel, Cangrelor | P2Y purinoceptor 12 | |||
| GPIIb/IIIa inhibitors | Tirofiban, Eptifibatide, Abciximb | platelet glycoprotein (GP) IIb/IIIa receptor | |||
| PAR-1 inhibitors | Vorapaxar | Proteinase-activated receptor 1 |
| Atherogenic Phase | Endothelial Cell-Derived Atheroprotective Effects | |
|---|---|---|
| EZH2 Antagonism | SIRT1 Agonism | |
| Endothelial cell activation, dysfunction, and fatty streak formation |
| |
| Inflammatory cell infiltration and inflammation |
|
|
| Neointima formation |
|
|
| Atherothrombosis |
| |
| Mechanism | Drug | Field of Use | Clinical Phase of Development | Number of Active Studies | Developer | Clinical Trial Identifier (s) |
|---|---|---|---|---|---|---|
| EZH2 antagonist | CPI-0209 | Oncology | I–II | 1 | Constellation Pharmaceuticals | NCT04104776 |
| CPI-1205 | Oncology | I–II | 3 | Constellation Pharmaceuticals | NCT02395601, NCT03525795, NCT03480646 | |
| GSK2816126 | Oncology | I | 1 | GlaxoSmithKline | NCT02082977 | |
| HH2853 | Oncology | I | 1 | Haihe Pharmaceutical | NCT04390737 | |
| PF-06821497 | Oncology | I | 1 | Pfizer | NCT03460977 | |
| SHR2554 | Oncology | I–II | 5 | Jiangsu HengRui Medicine | NCT04577885, NCT04627129, NCT04335266, NCT03741712, NCT04407741 | |
| MAK683 | I | 1 | Novartis | NCT02900651 | ||
| Tazemetostat | Oncology | I–III | 15 | Epizyme | NCT03009344, NCT03010982, NCT03028103, NCT02220842, NCT02875548, NCT03456726, NCT01897571, NCT02889523, NCT03155620, NCT04204941, NCT04224493, NCT02860286, NCT04179864, NCT02601950, NCT03854474 | |
| SIRT1 agonist | Quercetin | Cardiovascular | II–III | 3 | Quercegen Pharmaceuticals, Boehringer Ingelheim | NCT03943459, NCT02195232, NCT02191280 |
| Orthopaedics | III | 1 | Nestlé | NCT00330096 | ||
| Pulmonology | I–II | 3 | Quercegen Pharmaceuticals, AB Science | NCT03989271, NCT01708278, NCT04622865 | ||
| Resveratrol | Cardiovascular | I–III | 6 | Atrium Innovations, DSM Nutritional Products, Gateway Health Alliance, KGK Science, Fluxome Sciences | NCT01964846, NCT01364961, NCT01564381, NCT02415114, NCT01914081, NCT01668836 | |
| Dermatology | I | 1 | TCI Co | NCT04456829 | ||
| Metabolic | II–III | 9 | DSM Nutritional Products, Vedic Lifesciences | NCT01038089, NCT02216552, NCT02129595, NCT02565979, NCT01635114, NCT00823381, NCT00998504, NCT02834078, NCT02219906 | ||
| Neurology | I–II | 9 | Bial-Portela (BIA 6-512), Evolva (Veri-te) | NCT03095092, NCT03093389, NCT03095105, NCT03091543, NCT03094156, NCT03097211, NCT04314739, NCT03448094, NCT02621554 | ||
| Nephrology | III | 1 | NCT02433925 | |||
| Oncology | I | GlaxoSmithKline | NCT00920803 | |||
| Pulmonology | I–III | 1 | DSM Nutritional Products (Resvida) | NCT02245932, NCT02245962, NCT04166396 | ||
| SRT2379 | Metabolic | I | 2 | GlaxoSmithKline | NCT01262911, NCT01416376 | |
| Nephrology | I | 1 | GlaxoSmithKline | NCT01018628 | ||
| SRT2104 | Metabolic | I–II | 7 | GlaxoSmithKline | NCT00938275, NCT00933530, NCT00933062, NCT00937872, NCT01031108, NCT00937326, NCT01018017 | |
| Nephrology | I | 1 | GlaxoSmithKline | NCT01014117 | ||
| Pulmonology | I | 1 | GlaxoSmithKline | NCT00920660 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fledderus, J.; Vanchin, B.; Rots, M.G.; Krenning, G. The Endothelium as a Target for Anti-Atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1. J. Pers. Med. 2021, 11, 103. https://doi.org/10.3390/jpm11020103
Fledderus J, Vanchin B, Rots MG, Krenning G. The Endothelium as a Target for Anti-Atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1. Journal of Personalized Medicine. 2021; 11(2):103. https://doi.org/10.3390/jpm11020103
Chicago/Turabian StyleFledderus, Jolien, Byambasuren Vanchin, Marianne G. Rots, and Guido Krenning. 2021. "The Endothelium as a Target for Anti-Atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1" Journal of Personalized Medicine 11, no. 2: 103. https://doi.org/10.3390/jpm11020103
APA StyleFledderus, J., Vanchin, B., Rots, M. G., & Krenning, G. (2021). The Endothelium as a Target for Anti-Atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1. Journal of Personalized Medicine, 11(2), 103. https://doi.org/10.3390/jpm11020103