
Development and Validation of Decision Rules Models to Stratify Coronary Artery Disease, Diabetes, and Hypertension Risk in Preventive Care: Cohort Study of Returning UK Biobank Participants

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Biomarkers, Physical Measurements and Polygenic Risk Scores
2.3. Coronary Artery Disease
2.4. Type 2 Diabetes
2.5. Hypertension
2.6. Polygenic Risk Scores
2.7. Ascertainment of Disease Incidence
2.8. Statistical Analysis
3. Results
3.1. Population Characteristics
3.2. Polygenic Risk Scores
3.3. Sensitivity Analysis
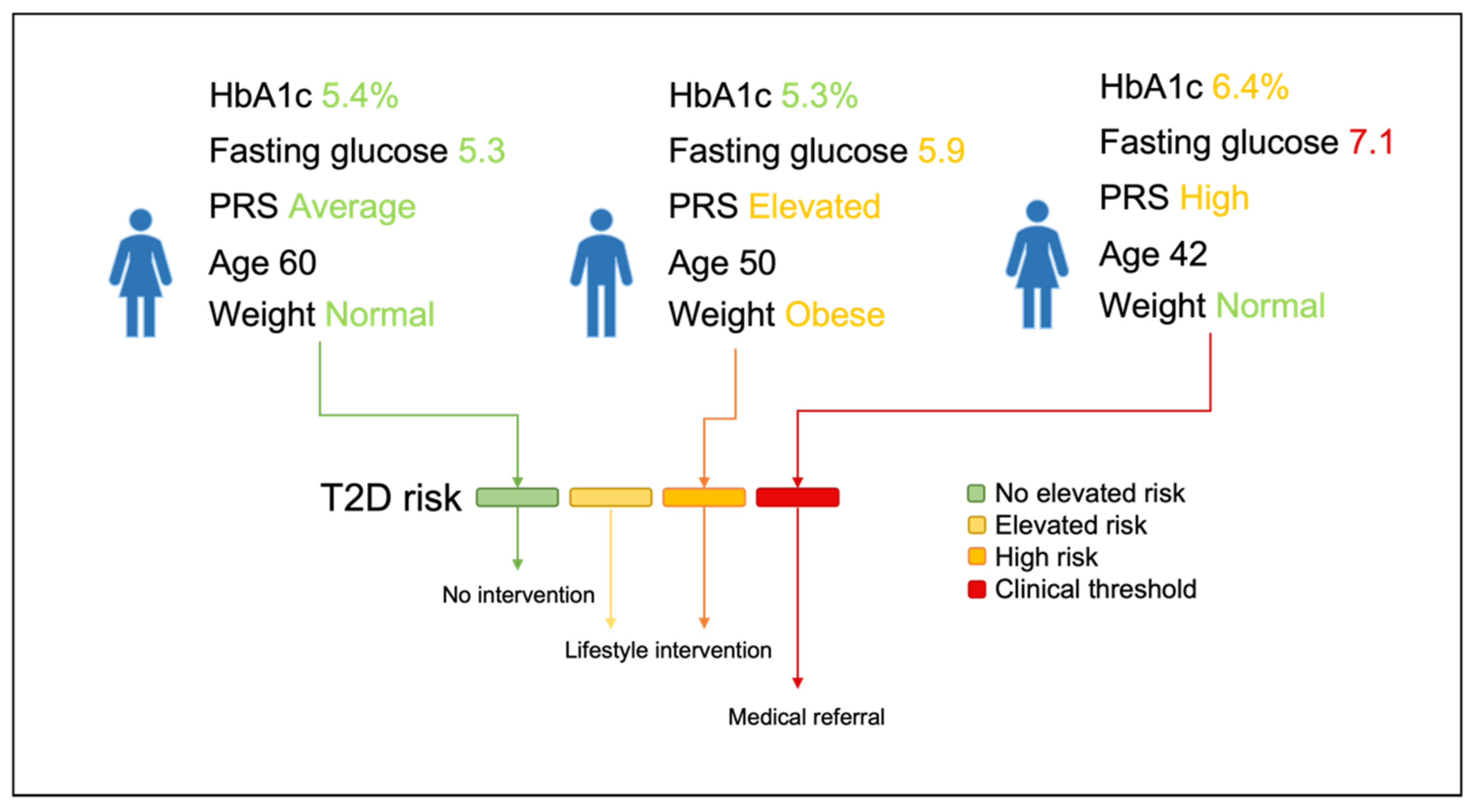
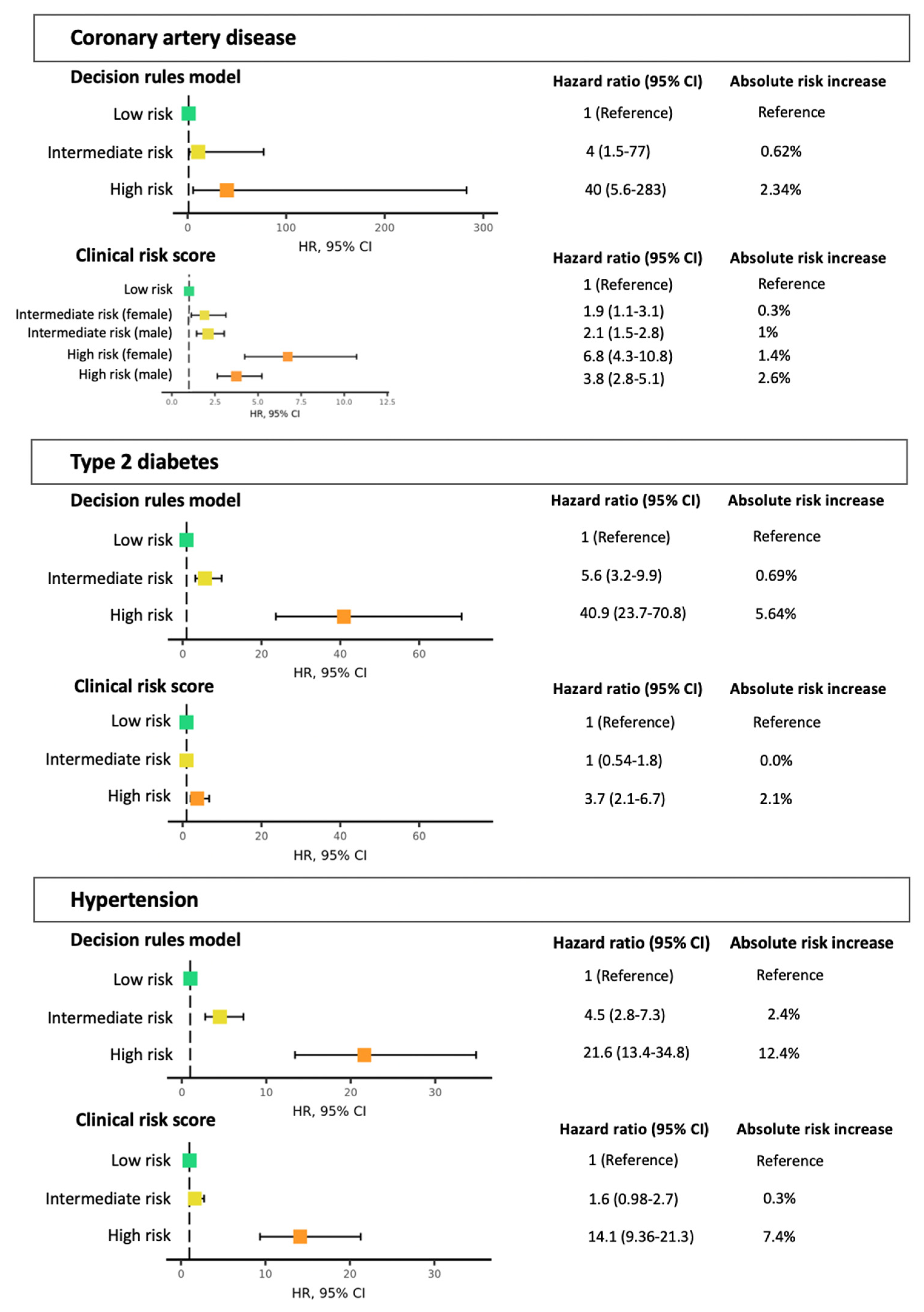
3.4. Risk Stratification and Lifestyle Advice Recommendations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Mortality Collaborators. Global, regional, and national age-specific mortality and life expectancy, 1950–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1684–1735. [Google Scholar] [CrossRef]
- WHO. Health Systems Performance Assessment: Debates, Methods and Empiricism. Available online: https://www.who.int/publications/2003/hspa/en/ (accessed on 30 October 2020).
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global Burden of Disease Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [PubMed]
- Franklin, B.A.; Cushman, M. Recent Advances in Preventive Cardiology and Lifestyle Medicine. Circulation 2011, 123, 2274–2283. [Google Scholar] [CrossRef]
- Magkos, F.; Hjorth, M.F.; Astrup, A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 545–555. [Google Scholar] [CrossRef]
- Wilson, P.W.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of coronary heart disease using risk factor categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef]
- Wilson, P.W.; Meigs, J.B.; Sullivan, L.; Fox, C.S.; Nathan, D.M.; D’Agostino, R.B. Prediction of incident diabetes mellitus in middle-aged adults: The Framingham Offspring Study. Arch. Intern. Med. 2007, 167, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.I.; Pencina, M.J.; Wang, T.J.; Benjamin, E.J.; Lanier, K.J.; Levy, D.; D’Agostino, R.B.; Kannel, W.B.; Vasan, R.S. A risk score for predicting near-term incidence of hypertension: The Framingham Heart Study. Ann. Intern. Med. 2008, 148, 102–110. [Google Scholar] [CrossRef]
- Damen, J.A.; Pajouheshnia, R.; Heus, P.; Moons, K.G.M.; Reitsma, J.B.; Scholten, R.J.P.M.; Hooft, L.; Debray, T.P.A. Performance of the Framingham risk models and pooled cohort equations for predicting 10-year risk of cardiovascular disease: A systematic review and meta-analysis. BMC Med. 2019, 17, 109. [Google Scholar] [CrossRef]
- Damen, J.A.; Hooft, L.; Schuit, E.; Debray, T.P.A.; Collins, G.S.; Tzoulaki, I.; Lassale, C.M.; Siontis, G.C.M.; Chiocchia, V.; Roberts, C.; et al. Prediction models for cardiovascular disease risk in the general population: Systematic review. BMJ 2016, 353, i2416. [Google Scholar] [CrossRef]
- Mars, N.J.; Koskela, J.T.; Ripatti, P.; Kiiskinen, T.T.J.; Havulinna, A.S.; Lindbohm, J.V.; Ahola-Olli, A.; Kurki, M.; Karjalainen, J.; Palta, P.; et al. Polygenic and clinical risk scores and their impact on age at onset of cardiometabolic diseases and common cancers. Nat. Med. 2020, 26, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Shi, J.; Garcia-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- Rutten-Jacobs, L.C.; Larsson, S.C.; Malik, R.; Rannikmäe, K.; MEGASTROKE Consortium; International Stroke Genetics Consortium; Sudlow, C.L.; Dichgans, M.; Markus, H.S.; Traylor, M. Genetic risk, incident stroke, and the benefits of adhering to a healthy lifestyle: Cohort study of 306 473 UK Biobank participants. BMJ 2018, 363, k4168. [Google Scholar] [CrossRef] [PubMed]
- Said, M.A.; Verweij, N.; van der Harst, P. Associations of Combined Genetic and Lifestyle Risks with Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol. 2018, 3, 693–702. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Himmelfarb, C.D.; Sondra, M.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar] [CrossRef] [PubMed]
- Nederlands Huisartsen Genootschap. Diabetes Mellitus Type 2, Derde Herziening. 2018. Available online: https://richtlijnen.nhg.org/standaarden/diabetes-mellitus-type-2#volledige-tekst-literatuur (accessed on 15 July 2021).
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Ann. Intern. Med. 2009, 15, 264–269. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [PubMed]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme High High-Density Lipoprotein Cholesterol Is Paradoxically Associated With High Mortality in Men and Women: Two Prospective Cohort Studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Third Report of the National Cholesterol Education Program (NCEP). Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Final Report. Circulation 2002, 106, 3143. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, M.; Khaw, K.-T.; Gudnason, V. Triglycerides and the Risk of Coronary Heart Disease: 10 158 Incident Cases among 262,525 Participants in 29 Western Prospective Studies. Circulation 2007, 115, 450–458. [Google Scholar] [CrossRef]
- Ikezaki, H.; Fisher, V.A.; Lim, E.; Ai, M.; Liu, C.T.; Adrienne Cupples, L.; Nakajima, K.; Asztalos, B.F.; Furusyo, N.; Schaefer, E.J. Direct Versus Calculated LDL Cholesterol and C-Reactive Protein in Cardiovascular Disease Risk Assessment in the Framingham Offspring Study. Clin. Chem. 2019, 65, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Penson, P.E.; Long, D.L.; Howard, G.; Toth, P.P.; Muntner, P.; Howard, V.J.; Safford, M.M.; Jones, S.R.; Martin, S.S.; Mazidi, M.; et al. Associations between very low concentrations of low-density lipoprotein cholesterol, high sensitivity C-reactive protein, and health outcomes in the Reasons for Geographical and Racial Differences in Stroke (REGARDS) study. Eur. Heart J. 2018, 39, 3641–3653. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Hemmingsen, B.; Metzendorf, M.-I.; Takwoingi, Y. Development of type 2 diabetes mellitus in people with intermediate hyperglycaemia. Cochrane Database Syst. Rev. 2018, 10, CD012661. [Google Scholar] [CrossRef]
- Ren, Y.; Luo, X.; Wang, C.; Yin, L.; Pang, C.; Feng, T. Prevalence of hypertriglyceridemic waist and association with risk of type 2 diabetes mellitus: A meta-analysis. Diabetes Metab. Res. Rev. 2016, 32, 405–412. [Google Scholar] [CrossRef]
- Riaz, H.; Khan, M.S.; Siddiqi, T.J.; Usman, M.S.; Shah, N.; Goyal, A.; Khan, S.S.; Mookadam, F.; Krasuski, R.A.; Ahmed, H. Association Between Obesity and Cardiovascular Outcomes: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. JAMA Netw. Open 2018, 1, e183788. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Hamaguchi, M.; Tanaka, M.; Obora, A.; Kojima, T.; Fukui, M. Metabolically healthy obesity without fatty liver and risk of incident type 2 diabetes: A meta-analysis of prospective cohort studies. Obes. Res. Clin. Pract. 2018, 12, 4–15. [Google Scholar] [CrossRef]
- Emdin, C.A.; Anderson, S.G.; Woodward, M.; Rahimi, K. Usual Blood Pressure and Risk of New-Onset Diabetes: Evidence from 4.1 Million Adults and a Meta-Analysis of Prospective Studies. J. Am. Coll. Cardiol. 2015, 66, 1552–1562. [Google Scholar] [CrossRef]
- Reboussin, D.M.; Allen, N.B.; Griswold, M.E.; Guallar, E.; Hong, Y.; Lackland, D.T.; MillerIII, E.R.; Polonsky, T.; Thompson-Paul, A.M.; Vupputuri, S. Systematic Review for the 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2018, 138, e595–e616. [Google Scholar] [CrossRef] [PubMed]
- Brunström, M.; Carlberg, B. Association of Blood Pressure Lowering With Mortality and Cardiovascular Disease across Blood Pressure Levels: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2018, 178, 28–36. [Google Scholar] [CrossRef]
- Sundström, J.; Arima, H.; Jackson, R.; Turnbull, F.; Rahimi, K.; Chalmers, J.; Woodward, M.; Neal, B.; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of blood pressure reduction in mild hypertension: A systematic review and meta-analysis. Ann. Intern. Med. 2015, 162, 184–191. [Google Scholar] [CrossRef]
- Thomopoulos, C.; Parati, G.; Zanchetti, A. Effects of blood pressure lowering on outcome incidence in hypertension: 2. Effects at different baseline and achieved blood pressure levels—Overview and meta-analyses of randomized trials. J. Hypertens. 2014, 32, 2296–2304. [Google Scholar] [CrossRef]
- Hong, Z.; Wu, T.; Zhou, S.; Huang, B.; Wang, J.; Jin, D.; Geng, D. Effects of anti-hypertensive treatment on major cardiovascular events in populations within prehypertensive levels: A systematic review and meta-analysis. J. Hum. Hypertens. 2018, 32, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Anna Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension (ESH). Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Yin, L.; Liu, W.; Liu, X.; Xiang, Q.; Qian, Z.; Xiang, Q.; Qian, Z.; Ma, J.; Chen, H.; et al. Associations of anthropometric adiposity indexes with hypertension risk: A systematic review and meta-analysis including PURE-China. Medicine 2018, 97, e13262. [Google Scholar] [CrossRef]
- Zho, Q.; Shi, Y.; Li, Y.-Q.; Ping, Z.; Wang, C.; Liu, X.; Lu, J.; Mao, Z.X.; Zhao, J.; Yin, L.; et al. Body mass index, abdominal fatness, and hypertension incidence: A dose-response meta-analysis of prospective studies. J. Hum. Hypertens. 2018, 32, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-C.; George, N.I.; Chang, C.-W.; Hicks, K.A. Assessing Sex Differences in the Risk of Cardiovascular Disease and Mortality per Increment in Systolic Blood Pressure: A Systematic Review and Meta-Analysis of Follow-Up Studies in the United States. PLoS ONE 2017, 12, e017021. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Vilhjálmsson, B.J.; Yang, J.; Finucane, H.K.; Gusev, A.; Lindström, S.; Ripke, S.; Genovese, G.; Loh, P.R.; Bhatia, G.; Do, R.; et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015, 97, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Willer, C.; Sanna, S.; Abecasis, G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009, 10, 387–406. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef]
- Pencina, M.J.; Demler, O.V. Novel metrics for evaluating improvement in discrimination: Net reclassification and integrated discrimination improvement for normal variables and nested models. Stat. Med. 2012, 31, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Pencina, M.J.; D’Agostino Sr, R.B.; D’Agostino, R.B., Jr.; Vasan, R.S. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat. Med. 2008, 27, 157–172. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Aulchenko, Y.S.; van Duijn, C.M.; Janssens, A.C. PredictABEL: An R package for the assessment of risk prediction models. Eur. J. Epidemiol. 2011, 26, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Lear, S.A.; James, P.T.; Ko, G.T.; Kumanyika, S. Appropriateness of waist circumference and waist-to-hip ratio cutoffs for different ethnic groups. Eur. J. Clin. Nutr. 2010, 64, 42–61. [Google Scholar] [CrossRef]
- Flack, J.M.; Adekola, B. Blood pressure and the new ACC/AHA hypertension guidelines. Trends Cardiovasc. Med. 2020, 30, 160–164. [Google Scholar] [CrossRef]
- D’Agostino, R.B.; Grundy, S.; Sullivan, L.M.; Wilson, P.; CHD Risk Prediction Group. Validation of the Framingham Coronary Heart Disease Prediction Scores. Results of a Multiple Ethnic Groups Investigation. JAMA 2001, 286, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Koller, M.T.; Leening, M.J.G.; Wolbers, M.; Steyerberg, E.W.; Hunink, M.; Schoop, R.; Hofman, A.; Bucher, H.C.; Psaty, B.M.; Lloyd-Jones, D.M.; et al. Development and Validation of a Coronary Risk Prediction Model for Older U.S. and European Persons in the Cardiovascular Health Study and the Rotterdam Study. Ann. Intern. Med. 2012, 157, 389–397. [Google Scholar] [CrossRef]
- Kivimäki, M.; Batty, G.D.; Singh-Manoux, A.; Ferrie, J.E.; Tabak, A.G.; Jokela, M.; Marmot, M.G.; Smith, G.D.; Shipley, M.J. Validating the Framingham Hypertension Risk Score: Results from the Whitehall II Study. Hypertension 2009, 54, 496–501. [Google Scholar] [CrossRef]
- Syllos, D.H.; Calsavara, V.F.; Bensenor, I.M.; Lotufo, P.A. Validating the Framingham Hypertension Rsk Score: A 4-year follow-up from the Brazilian Longitudinal Study of the Adult Health (ELSA-Brasil). J. Clin. Hypertens. 2020, 22, 850–856. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.; Wang, C.; Ren, Y.; Wang, B.; Zhang, L.; Yang, X.; Zhao, Y.; Han, C.; Pang, C.; et al. Development and Validation of a Risk-Score Model for Type 2 Diabetes: A Cohort Study of a Rural Adult Chinese Population. PLoS ONE 2016, 11, e0152054. [Google Scholar] [CrossRef]
- Carroll, S.J.; Paquet, C.; Howard, N.J.; Adams, R.J.; Taylor, A.W.; Daniel, M. Validation of continuous clinical indices of cardiometabolic risk in a cohort of Australian adults. BMC Cardiovasc. Disord. 2014, 14, 27. [Google Scholar] [CrossRef]
- Udler, M.S.; Kim, J.; von Grotthuss, M.; Bonàs-Guarch, S.; Cole, J.B.; Chiou, J.; Anderson, C.D.; Boehnke, M.; Laakso, M.; Atzmon, G.; et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 2018, 15, e1002654. [Google Scholar] [CrossRef]
- Ahlqvist, E.; Storm, P.; Karajamaki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Mansour Aly, D.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef]
- Lean, M.E.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): An open-label, cluster-randomised trial. Lancet 2018, 391, 541–551. [Google Scholar] [CrossRef]
- Kim, S.E.; Castro Sweet, C.M.; Cho, E.; Tsai, J.; Cousineau, M.R. Evaluation of a Digital Diabetes Prevention Program Adapted for Low-Income Patients, 2016–2018. Prev. Chronic Dis. 2019, 16, E155. [Google Scholar] [CrossRef] [PubMed]
- Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; McKenzie, A.L.; Bhanpuri, N.H.; Campbell, W.W.; Volek, J.S.; Phinney, S.D.; McCarter, J.P. Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Non-randomized Clinical Trial. Front. Endocrinol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017, 60, 793–799. [Google Scholar] [CrossRef]
- Levin, M.G.; Rader, D.J. Polygenic Risk Scores and Coronary Artery Disease: Ready for Prime Time? Circulation 2020, 141, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Müller-Riemenschneider, F.; Holmberg, C.; Rieckmann, N.; Kliems, H.; Rufer, V.; Müller-Nordhorn, J.; Willich, S.N. Barriers to Routine Risk-Score Use for Healthy Primary Care Patients: Survey and Qualitative Study. Arch. Intern. Med. 2010, 170, 719–724. [Google Scholar] [CrossRef]
- Kappen, T.H.; Van Loon, K.; Kappen, M.A.; van Wolfswinkel, L.; Vergouwe, Y.; van Klei, W.A.; Moons, K.G.; Kalkman, C.J. Barriers and facilitators perceived by physicians when using prediction models in practice. J. Clin. Epidemiol. 2016, 70, 136–145. [Google Scholar] [CrossRef][Green Version]
- Rosselo, X.; Dorresteijn, J.A.N.; Janssen, A.; Lambrinou, E.; Scherrenberg, M.; Bonnefoy-Cudraz, E.; Cobain, M.; Piepoli, M.F.; Visseren, F.L.; Dendale, P. Risk prediction tools in cardiovascular disease prevention. Eur. J. Prev. Cardiol. 2019, 26, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.M.; Leip, E.P.; Larson, M.G.; D’Agostino, R.B.; Beiser, A.; Wilson, P.W.F.; Wolf, P.A.; Levy, D. Prediction of Lifetime Risk for Cardiovascular Disease by Risk Factor Burden at 50 Years of Age. Circulation 2006, 113, 791–798. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, H.; et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 2021, 53, 420–425. [Google Scholar] [CrossRef]
- Liu, Q.; Davis, J.; Han, X.; Mackey, D.A.; MacGregor, S.; Craig, J.E.; Si, L.; Hewitt, A.W. Cost-effectiveness of polygenic risk profiling for primary open-angle glaucoma in the United Kingdom and Australia. medRxiv 2021. [Google Scholar] [CrossRef]
- Wong, J.; Chai, J.H.; Yeoh, Y.S.; Mohamed Riza, N.K.; Liu, J.; Teo, Y.-Y. Cost effectiveness analysis of a polygenic risk tailored breast cancer screening programme in Singapore. BMC Health Serv. Res. 2021, 21, 379. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pennells, L.; Kaptoge, S.; Nelson, C.P.; Ritchie, S.C.; Abraham, G.; Arnold, M.; Bell, S.; Bolton, T.; Burgess, S.; et al. Polygenic risk scores in cardiovascular risk prediction: A cohort study and modelling analyses. PLoS Med. 2021, 18, e1003498. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Bodinier, B.; Bond, T.A.; Chadeau-Hyam, M.; Evangelou, E.; Moons, K.G.M.; Dehghan, A.; Muller, D.C.; Elliott, P.; Tzoulaki, I. Predictive Accuracy of a Polygenic Risk Score–Enhanced Prediction Model vs. a Clinical Risk Score for Coronary Artery Disease. JAMA 2020, 323, 636–645. [Google Scholar] [CrossRef]
- Sud, A.; Turnbull, C.; Houlston, R. Will polygenic risk scores for cancer ever be clinically useful? NPJ Precis. Oncol. 2021, 5, 40. [Google Scholar] [CrossRef]
- Huckvale, K.; Jason Wang, C.; Majeed, A.; Car, J. Digital health at fifteen: More human (more needed). BMC Med. 2019, 17, 62. [Google Scholar] [CrossRef]
- Gordon, W.J.; Landman, A.; Zhang, H.; Bates, D.W. Beyond validation: Getting health apps into clinical practice. NPJ Digit. Med. 2020, 3, 14. [Google Scholar] [CrossRef]
- Natanson, E. Healthcare Apps: A Boon, Today and Tomorrow. Forbes. Available online: https://www.forbes.com/sites/eladnatanson/2020/07/21/healthcare-apps-a-boon-today-and-tomorrow/?sh=59bfd4ab1bb9 (accessed on 10 July 2021).
- Natarajan, P.; Young, R.; Stitziel, N.O.; Padmanabhan, S.; Baber, U.; Mehran, R.; Sartori, S.; Fuster, V.; Reilly, D.F.; Butterworth, A.; et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 2017, 135, 2091–2101. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Stitziel, N.O.; Smith, J.G.; Chasman, D.I.; Caulfield, M.; Devlin, J.J.; Nordio, F.; Hyde, C.; Cannon, C.P.; Sacks, F.; et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet 2015, 385, 2264–2271. [Google Scholar] [CrossRef]
- Fry, A.; Littlejohns, T.J.; Sudlow, C.; Doherty, N.; Adamska, A.; Sprosen, T.; Collins, R.; Allen, N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants with Those of the General Population. Am. J. Epidemiol. 2017, 186, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.B.; Mathews, S.C.; Dorsey, E.R.; Bates, D.W.; Safavi, K. Direct-to-consumer digital health. Lancet Digit. Health 2020, 2, e163–e165. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Mean (SD), Percentage (%), or Number of Participants (N) |
|---|---|
| Total, No. | 60,782 |
| Age, y | 56.3 (7.59) |
| Female | 51.2% |
| With CAD at follow-up | n = 500 |
| With diabetes at follow-up | n = 1005 |
| With hypertension at follow-up | n = 2379 |
| Blood pressure, mm Hg | |
| Systolic | 138 (19) |
| Diastolic | 82 (10) |
| Smoking | |
| Ideal (never or stopped >1 y ago) | 82.7% |
| Intermediate (stopped <1 y ago) | 0.3% |
| Current smoker | 5.6% |
| Body composition | |
| BMI | 26.8 (4.35) |
| Waist circumference (cm) | 88.7 (12.8) |
| Waist-to-hip ratio | 0.86 (0.09) |
| Body fat percentage (%) | 30.2 (8.3) |
| Blood biomarkers | |
| Total cholesterol (mmol/L) | 5.71 (1.1) |
| LDL cholesterol (mmol/L) | 3.58 (0.84) |
| HDL cholesterol (mmol/L) | 1.47 (0.38) |
| Triglycerides (mmol/L) | 1.68 (0.97) |
| hs-CRP (mg/L) | 2.17 (3.8) |
| Fasting glucose (mmol/L) | 5.0 (1.0) |
| HbA1c (mmol/mmol) | 35.2 (5.3) |
| Albumin-creatinine ratio | 2.4 (8.3) |
| Family history | |
| No family history of diabetes | 83.2% |
| Family history of diabetes (1 parent) | 16.8% |
| Family history of diabetes (both) | 0.01% |
| No family history of CAD | 58.1% |
| Family history of CAD (1 parent) | 41.8% |
| Family history of CAD (both) | 0.07% |
| No family history of hypertension | 57.3% |
| Family history of hypertension (1 parent) | 42.7% |
| Family history of hypertension (both) | 0.08% |
| Health Condition | Unadjusted PRS | PRS Adjusted for 4 PCs and Array Type | PRS Adjusted for 4 PCs, Array Type, Sex and Age | Age and Sex |
|---|---|---|---|---|
| CAD | 1.66 (0.93–2.35) | 2.25 (1.39–3.11) | 4.43 (3.14–5.74) | 2.55 (1.63–3.47) |
| T2D | 1.86 (1.33–2.39) | 2.61 (1.96–3.26) | 2.81 (2.12–3.50) | 1.47 (1.00–1.94) |
| HT | 1.37 (1.06–1.62) | 1.61 (1.30–1.92) | 1.77 (1.45–2.09) | 1.50 (1.21–1.79) |
| Model/Health Condition | Low Risk (N) | Low Risk Who Developed Disease (N) | Advised Lifestyle (N) | Advised Lifestyle Who Developed Disease (N) | AUROC (95% CI) |
|---|---|---|---|---|---|
| CAD (n = 21,969 women, 167 cases; 14,944 men, 333 cases) | |||||
| FRS women | 7426 | 22 | 5680 | 99 | 0.67 (0.63–0.71) |
| FRS men | 5171 | 55 | 4431 | 165 | 0.60 (0.58–0.63) |
| PRS | 25,839 | 173 | 3692 | 165 | 0.62 (0.60–0.64) |
| Rule model | 1521 | 0 $ | 14,980 | 360 | 0.66 (0.64–0.68) |
| Diabetes (n = 42,978, 1005 cases) | |||||
| FRS | 12,305 | 39 | 12,634 | 726 | 0.72 (0.71–0.73) |
| PRS | 30,084 | 467 | 4298 | 239 | 0.57 (0.56–0.58) |
| Rule model | 8351 | 13 | 14,169 | 819 | 0.75 (0.74–0.76) |
| Hypertension (n = 33,541, 2379 cases) | |||||
| FRS | 3359 | 23 | 26,587 | 2317 | 0.60 (0.59–0.60) |
| PRS | 23,479 | 1327 | 3354 | 391 | 0.54 (0.53–0.54) |
| Rule model | 2274 | 17 | 12,506 | 1759 | 0.70 (0.69–0.71) |
| Model/ Health Condition | Event | Non-Event | ||||
|---|---|---|---|---|---|---|
| CAD women (n = 21,969,167 cases) | ||||||
| Rules model | Rules model | |||||
| Framingham | Rec. intervention | Low-risk | Corr. reclass. (%) | Rec. intervention | Low-risk | Corr. reclass. (%) |
| Rec. intervention | 82 | 17 | 17% | 3746 | 1835 | 33% |
| Low risk | 35 | 33 | 51% | 4010 | 12,211 | 25% |
| CAD men (n = 14,944,333 cases) | ||||||
| Rules model | Rules model | |||||
| Framingham | Rec. intervention | Low-risk | Corr. reclass. (%) | Rec. intervention | Low-risk | Corr. reclass. (%) |
| Rec. intervention | 125 | 40 | 24% | 2558 | 1708 | 40% |
| Low-risk | 118 | 50 | 70% | 4306 | 6039 | 42% |
| T2D (n = 42,978, 1005 cases) | ||||||
| Rules model | Rules model | |||||
| Framingham | Rec. intervention | Low-risk | Corr. reclass. (%) | Rec. intervention | Low-risk | Corr. reclass. (%) |
| Rec. intervention | 617 | 109 | 15% | 6183 | 5725 | 48% |
| Low-risk | 202 | 77 | 72% | 7167 | 22,898 | 24% |
| Hypertension (n = 33,541, 2379 cases) | ||||||
| Rules model | Rules model | |||||
| Framingham | Rec. intervention | Low-risk | Corr. reclass. (%) | Rec. intervention | Low-risk | Corr. reclass. (%) |
| Rec. intervention | 1751 | 566 | 24% | 10,477 | 13,793 | 57% |
| Low-risk | 8 | 54 | 13% | 270 | 6622 | 4% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castela Forte, J.; Folkertsma, P.; Gannamani, R.; Kumaraswamy, S.; Mount, S.; de Koning, T.J.; van Dam, S.; Wolffenbuttel, B.H.R. Development and Validation of Decision Rules Models to Stratify Coronary Artery Disease, Diabetes, and Hypertension Risk in Preventive Care: Cohort Study of Returning UK Biobank Participants. J. Pers. Med. 2021, 11, 1322. https://doi.org/10.3390/jpm11121322
Castela Forte J, Folkertsma P, Gannamani R, Kumaraswamy S, Mount S, de Koning TJ, van Dam S, Wolffenbuttel BHR. Development and Validation of Decision Rules Models to Stratify Coronary Artery Disease, Diabetes, and Hypertension Risk in Preventive Care: Cohort Study of Returning UK Biobank Participants. Journal of Personalized Medicine. 2021; 11(12):1322. https://doi.org/10.3390/jpm11121322
Chicago/Turabian StyleCastela Forte, José, Pytrik Folkertsma, Rahul Gannamani, Sridhar Kumaraswamy, Sarah Mount, Tom J. de Koning, Sipko van Dam, and Bruce H. R. Wolffenbuttel. 2021. "Development and Validation of Decision Rules Models to Stratify Coronary Artery Disease, Diabetes, and Hypertension Risk in Preventive Care: Cohort Study of Returning UK Biobank Participants" Journal of Personalized Medicine 11, no. 12: 1322. https://doi.org/10.3390/jpm11121322
APA StyleCastela Forte, J., Folkertsma, P., Gannamani, R., Kumaraswamy, S., Mount, S., de Koning, T. J., van Dam, S., & Wolffenbuttel, B. H. R. (2021). Development and Validation of Decision Rules Models to Stratify Coronary Artery Disease, Diabetes, and Hypertension Risk in Preventive Care: Cohort Study of Returning UK Biobank Participants. Journal of Personalized Medicine, 11(12), 1322. https://doi.org/10.3390/jpm11121322