
Evidence for the Association between the Intronic Haplotypes of Ionotropic Glutamate Receptors and First-Episode Schizophrenia

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. SCH Patients/Control Subjects
2.2. Genomic DNA Analysis
2.3. Sequencing Data Analysis
2.4. Homology Modeling
2.5. Statistical Analysis
2.6. Segmentation of the Brain and Estimation of the BrainAGE Index
3. Results
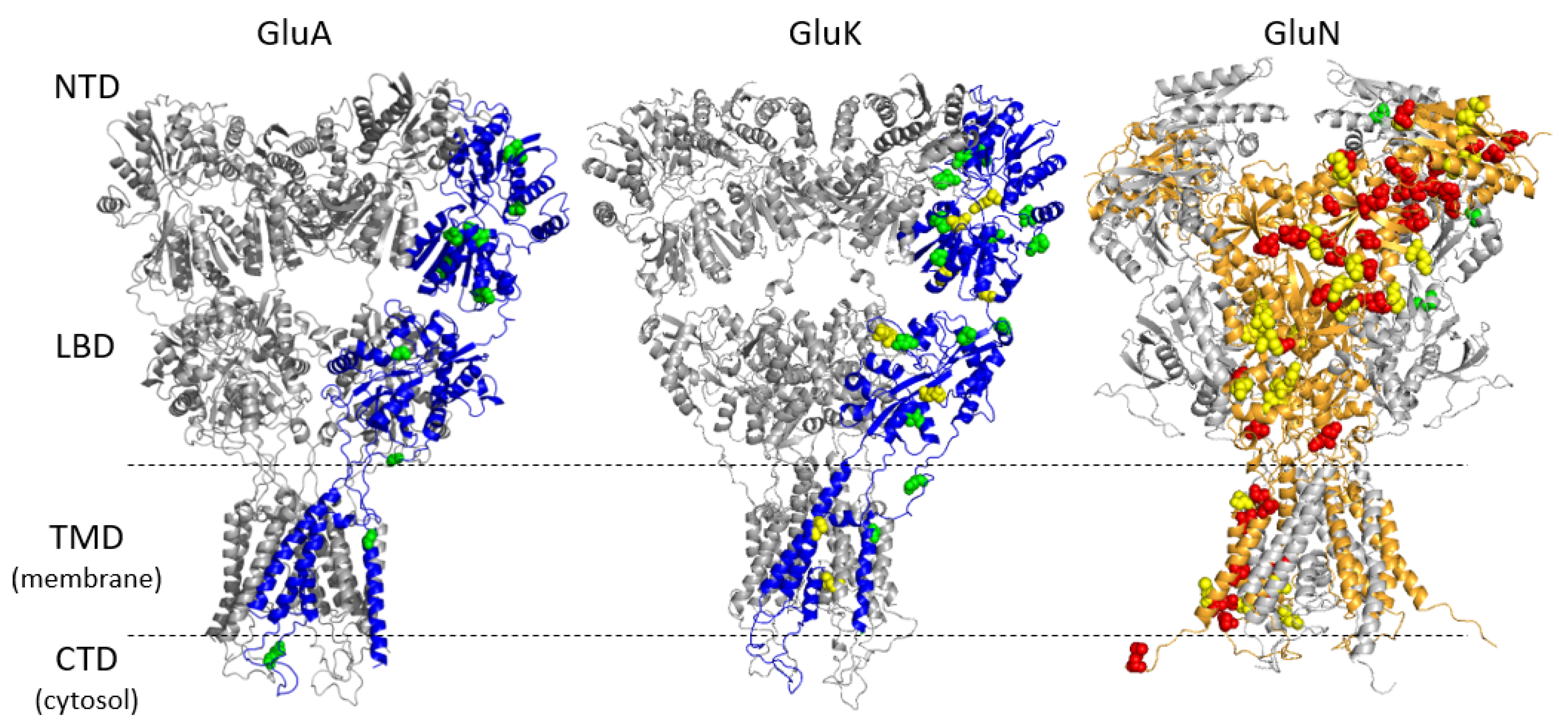
3.1. Genetic Variations in iGluR Genes
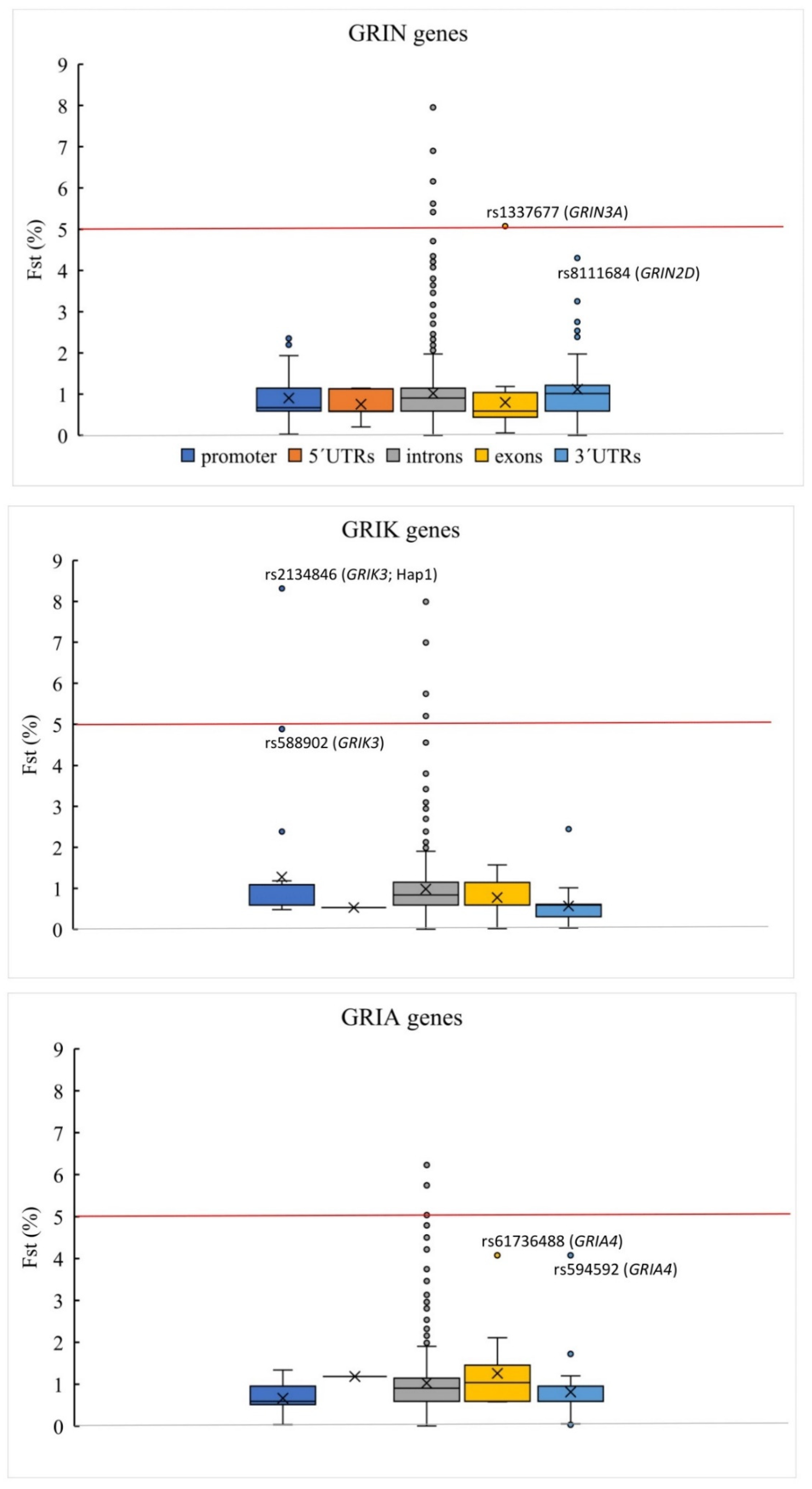
3.2. Genetic Variation Associated with FES
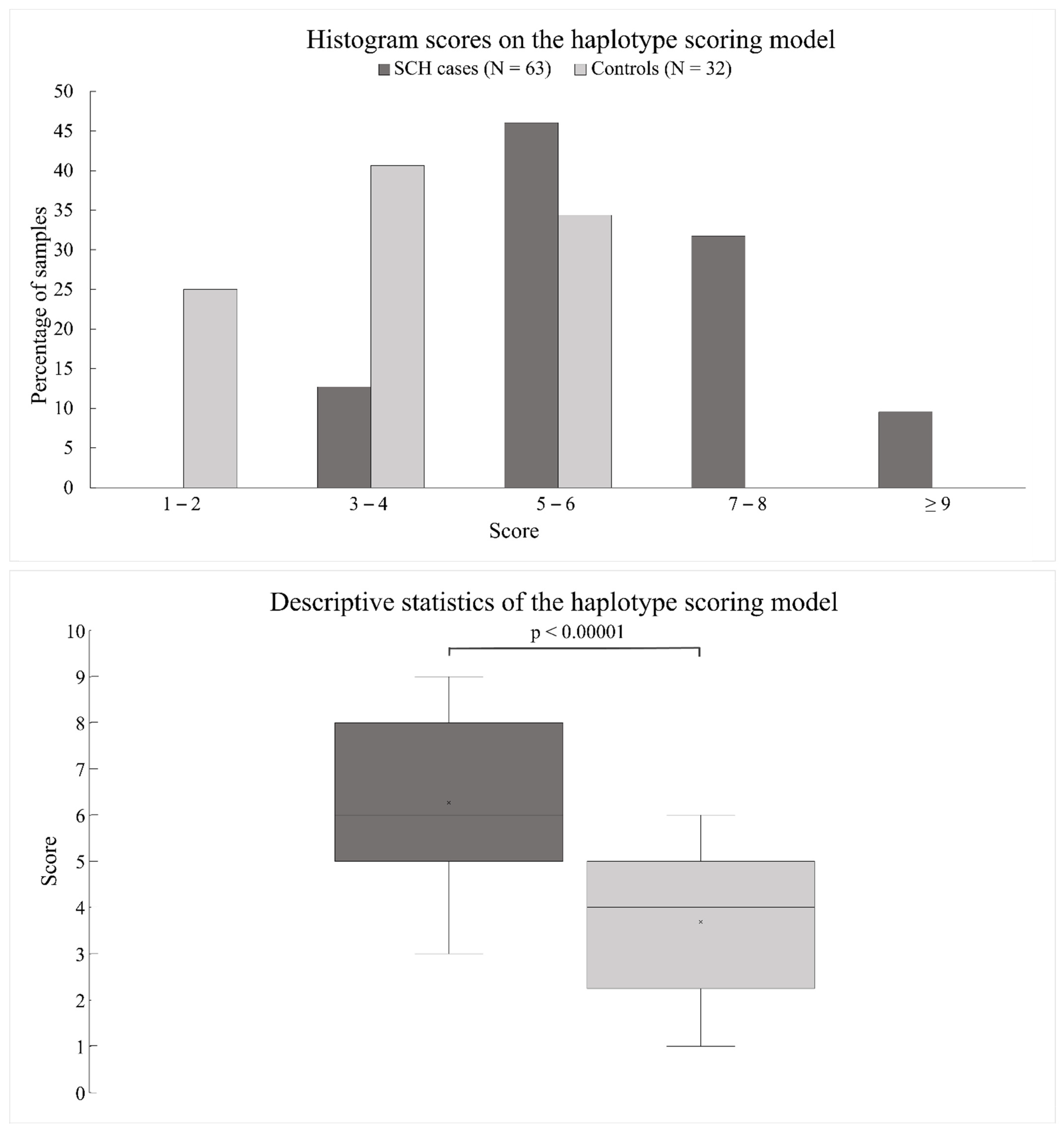
3.3. Scoring Model for the Estimation of Cumulative Risk
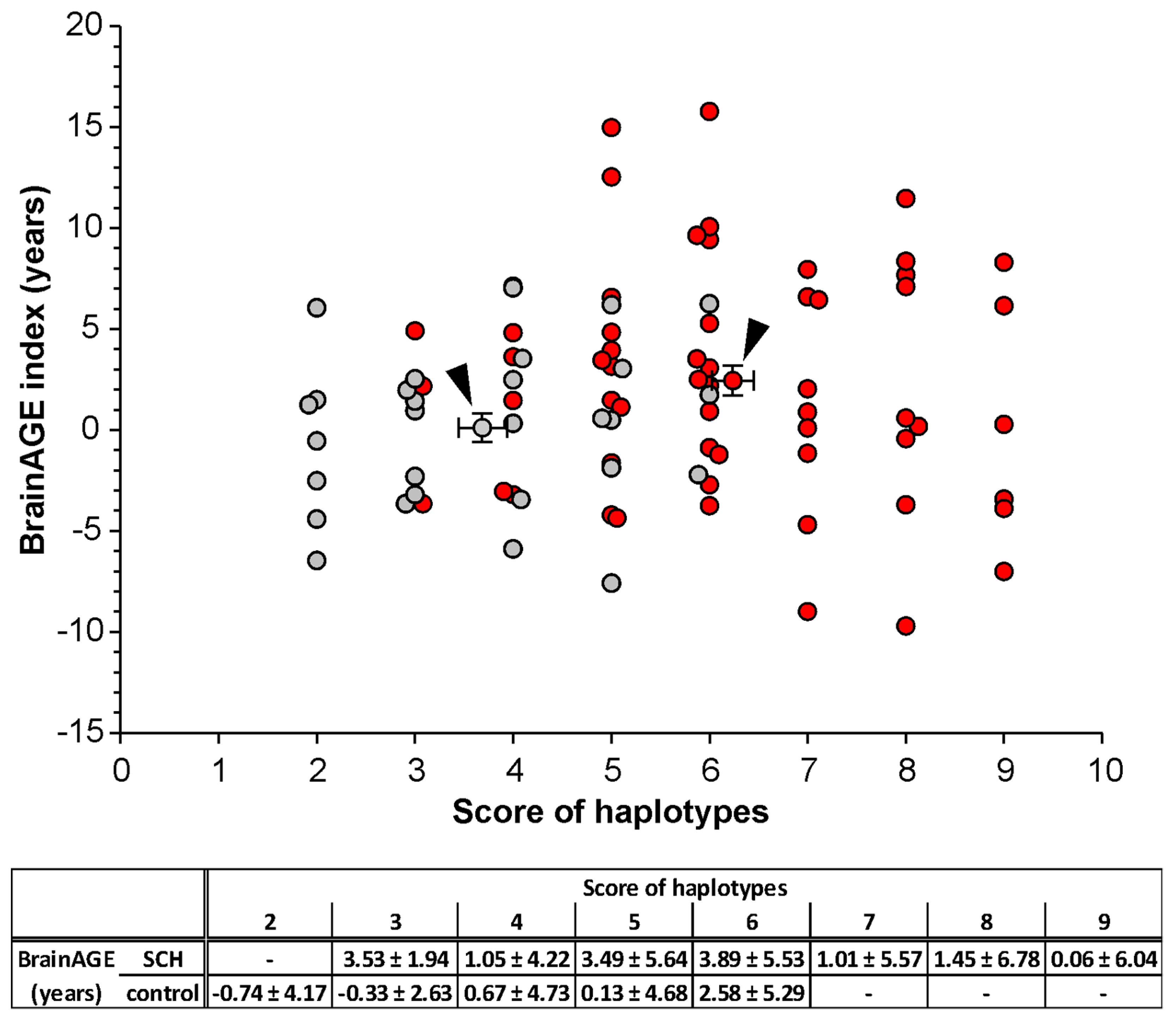
3.4. Correlation of the BrainAGE Index with the Haplotype Scoring Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, S.; Chant, D.; Welham, J.; McGrath, J. A Systematic Review of the Prevalence of Schizophrenia. PLoS Med. 2005, 2, e141. [Google Scholar] [CrossRef] [PubMed]
- Simeone, J.C.; Ward, A.J.; Rotella, P.; Collins, J.; Windisch, R. An evaluation of variation in published estimates of schizophrenia prevalence from 1990─2013: A systematic literature review. BMC Psychiatry 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Shih, R.A.; Belmonte, P.L.; Zandi, P.P. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int. Rev. Psychiatry 2004, 16, 260–283. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.E.; David, A.; Gold, J.M. Neurocognitive Deficits in Schizophrenia. In Schizophrenia; Blackwell Science Ltd.: Oxford, UK, 2003; pp. 168–184. [Google Scholar]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef]
- Kristiansen, L.; Huerta, I.; Beneyto, M.; Meador-Woodruff, J.H. NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 2007, 7, 48–55. [Google Scholar] [CrossRef]
- E McCullumsmith, R.; Hammond, J.H.; Shan, D.; Meador-Woodruff, J.H. Postmortem Brain: An Underutilized Substrate for Studying Severe Mental Illness. Neuropsychopharmacology 2013, 39, 65–87. [Google Scholar] [CrossRef]
- Weickert, C.S.; Fung, S.J.; Catts, V.S.; Schofield, P.; Allen, K.M.; Moore, L.; Newell, K.; Pellen, D.; Huang, X.-F.; Catts, S.V.; et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2012, 18, 1185–1192. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; Mcbain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef]
- Greger, I.H.; Mayer, M.L. Structural biology of glutamate receptor ion channels: Towards an understanding of mechanism. Curr. Opin. Struct. Biol. 2019, 57, 185–195. [Google Scholar] [CrossRef]
- Myers, S.J.; Dingledine, R.; Borges, K. Genetic Regulation of Glutamate Receptor Ion Channels. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Salussolia, C.; Wollmuth, L.P.; Scientist, M.; Program, T.; Brook, S. Assembly of AMPA receptors: Mechanisms and regulation. J. Physiol. 2014, 593, 39–48. [Google Scholar] [CrossRef]
- Greger, I.H.; Watson, J.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Elgersma, Y.; Silva, A.J. Molecular mechanisms of synaptic plasticity and memory. Curr. Opin. Neurobiol. 1999, 9, 209–213. [Google Scholar] [CrossRef]
- Hunt, D.; E Castillo, P. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef]
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental disorders. Lancet Psychiatry 2017, 4, 339–346. [Google Scholar] [CrossRef]
- Luby, E.D. Study of a New Schizophrenomimetic Drug—Sernyl. Arch. Neurol. Psychiatry 1959, 81, 363–369. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-Induced Exacerbation of Psychotic Symptoms and Cognitive Impairment in Neuroleptic-Free Schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with Reduced NMDA Receptor Expression Display Behaviors Related to Schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef]
- Miyatake, R.; Furukawa, A.; Suwaki, H. Identification of a novel variant of the human NR2B gene promoter region and its possible association with schizophrenia. Mol. Psychiatry 2002, 7, 1101–1106. [Google Scholar] [CrossRef][Green Version]
- Qin, S.; Zhao, X.; Pan, Y.; Liu, J.; Feng, G.; Fu, J.; Bao, J.; Zhang, Z.; He, L. An association study of the N-methyl-D-aspartate receptor NR1 subunit gene (GRIN1) and NR2B subunit gene (GRIN2B) in schizophrenia with universal DNA microarray. Eur. J. Hum. Genet. 2005, 13, 807–814. [Google Scholar] [CrossRef][Green Version]
- Rice, S.R.; Niu, N.; Berman, D.B.; Heston, L.L.; Sobell, J.L. Identification of single nucleotide polymorphisms (SNPs) and other sequence changes and estimation of nucleotide diversity in coding and flanking regions of the NMDAR1 receptor gene in schizophrenic patients. Mol. Psychiatry 2001, 6, 274–284. [Google Scholar] [CrossRef]
- Sakurai, K.; Toru, M.; Yamakawa-Kobayashi, K.; Arinami, T. Mutation analysis of the N-methyl-d-aspartate receptor NR1 subunit gene (GRIN1) in schizophrenia. Neurosci. Lett. 2000, 296, 168–170. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Liao, D.-L.; Chen, J.-Y.; Wang, Y.-C.; Lai, I.-C.; Liou, Y.-J.; Chen, Y.-J.; Luu, S.-U.; Chen, C.-H. Exomic sequencing of the glutamate receptor, ionotropic, N-methyl-d-aspartate 3A gene (GRIN3A) reveals no association with schizophrenia. Schizophr. Res. 2009, 114, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Iwayama, Y.; Fukuo, Y.; Ikeda, M.; Okochi, T.; Maekawa, M.; Toyota, T.; Yamada, K.; Hattori, E.; Ohnishi, T.; et al. A Population-Specific Uncommon Variant in GRIN3A Associated with Schizophrenia. Biol. Psychiatry 2013, 73, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.M.; Bowen, T.; Spurlock, G.; Norton, N.; Williams, H.; Hoogendoorn, B.; Owen, M.J.; O’Donovan, M.C. Determination of the genomic structure and mutation screening in schizophrenic individuals for five subunits of the N-methyl-D-aspartate glutamate receptor. Mol. Psychiatry 2002, 7, 508–514. [Google Scholar] [CrossRef][Green Version]
- Yu, Y.; Lin, Y.; Takasaki, Y.; Wang, C.; Kimura, H.; Xing, J.; Ishizuka, K.; Toyama, M.; Kushima, I.; Mori, D.; et al. Rare loss of function mutations in N-methyl-d-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, É.; Millet, B.; Fathalli, F.; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, M.; Yamada, K.; Yoshitsugu, K.; Toyota, T.; Suga, T.; Ohba, H.; Watanabe, A.; Hattori, E.; Shimizu, H.; Kumakura, T.; et al. A microsatellite repeat in the promoter of the N-methyl-d-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 2003, 13, 271–278. [Google Scholar] [CrossRef]
- Iwayama-Shigeno, Y.; Yamada, K.; Itokawa, M.; Toyota, T.; Meerabux, J.M.; Minabe, Y.; Mori, N.; Inada, T.; Yoshikawa, T. Extended analyses support the association of a functional (GT)n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci. Lett. 2005, 378, 102–105. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- International, T.; Consortium, S. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nat. Cell Biol. 2009, 460, 748–752. [Google Scholar] [CrossRef]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.; Kähler, A.K.; Akterin, S.; Bergen, S.; Collins, A.L.; Crowley, J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef]
- Frank, R.; McRae, A.F.; Pocklington, A.J.; Van De Lagemaat, L.N.; Navarro, P.; Croning, M.D.R.; Komiyama, N.H.; Bradley, S.J.; Challiss, R.A.J.; Armstrong, J.D.; et al. Clustered Coding Variants in the Glutamate Receptor Complexes of Individuals with Schizophrenia and Bipolar Disorder. PLoS ONE 2011, 6, e19011. [Google Scholar] [CrossRef]
- Kirov, G.; Pocklington, A.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2011, 17, 142–153. [Google Scholar] [CrossRef]
- Pocklington, A.J.; Rees, E.; Donovan, M.C.O.; Owen, M.J.; Pocklington, A.J.; Rees, E.; Walters, J.T.R.; Han, J.; Kavanagh, D.H.; Chambert, K.D. Novel Findings from CNVs Implicate Inhibitory and Excitatory Signaling Complexes in Schizophrenia Article Novel Findings from CNVs Implicate Inhibitory and Excitatory Signaling Complexes in Schizophrenia. Neuron 2015, 86, 1203–1214. [Google Scholar] [CrossRef]
- Purcell, S.M.; Moran, J.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.V.; Lecrubier, Y.; Sheehan, K.H.; Amorim, P.; Janavs, J.; Weiller, E.; Hergueta, T.; Baker, R.; Dunbar, G.C. The Mini-International Neuropsychiatric Interview (M.I.N.I.): The development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J. Clin. Psychiatry 1998, 59, 22–33; quiz 34–57. [Google Scholar] [PubMed]
- Kay, S.R.; Fiszbein, A.; Opler, L.A. The Positive and Negative Syndrome Scale (PANSS) for Schizophrenia. Schizophr. Bull. 1987, 13, 261–276. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Fritz, M.H.-Y.; O Korbel, J.; Benes, V. Alfred: Interactive multi-sample BAM alignment statistics, feature counting and feature annotation for long- and short-read sequencing. Bioinformatics 2019, 35, 2489–2491. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Zhang, Z.; Xin, D.; Wang, P.; Zhou, L.; Hu, L.; Kong, X.; Hurst, L.D. Noisy splicing, more than expression regulation, explains why some exons are subject to nonsense-mediated mRNA decay. BMC Biol. 2009, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population structure by f-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Franke, K.; Ziegler, G.; Klöppel, S.; Gaser, C. Estimating the age of healthy subjects from T1-weighted MRI scans using kernel methods: Exploring the influence of various parameters. NeuroImage 2010, 50, 883–892. [Google Scholar] [CrossRef]
- Franke, K.; Luders, E.; May, A.; Wilke, M.; Gaser, C. Brain maturation: Predicting individual BrainAGE in children and adolescents using structural MRI. NeuroImage 2012, 63, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- García-Recio, A.; Santos-Gómez, A.; Soto, D.; Julia-Palacios, N.; García-Cazorla, À.; Altafaj, X.; Olivella, M. GRIN database: A unified and manually curated repertoire of GRIN variants. Hum. Mutat. 2021, 42, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Hornig, T.; Grüning, B.; Kundu, K.; Houwaart, T.; Backofen, R.; Biber, K.; Normann, C. GRIN3B missense mutation as an inherited risk factor for schizophrenia: Whole-exome sequencing in a family with a familiar history of psychotic disorders. Genet. Res. 2017, 99, e1. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, H.; Ohi, K.; Hashimoto, R.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Yano-Umeda, S.; Saneyoshi, T.; Takeda, M.; Hayashi, Y. A Naturally Occurring Null Variant of the NMDA Type Glutamate Receptor NR3B Subunit Is a Risk Factor of Schizophrenia. PLoS ONE 2015, 10, e0116319. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.-L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, reviews0004.1. [Google Scholar] [CrossRef]
- Hu, Z.; Bruno, A.E. The Influence of 3′UTRs on MicroRNA Function Inferred from Human SNP Data. Comp. Funct. Genom. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, M.; Wang, Q.; He, G.; Fu, Y.; Li, H.; Yu, S. Polymorphisms in MicroRNA Genes And Genes Involving in NMDAR Signaling and Schizophrenia: A Case-Control Study in Chinese Han Population. Sci. Rep. 2015, 5, 12984. [Google Scholar] [CrossRef]
- Shen, H.; Li, Z. miRNAs in NMDA receptor-dependent synaptic plasticity and psychiatric disorders. Clin. Sci. 2016, 130, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, K.N.; Wu, C.C.-C.; Sinha, N.; Loll-Krippleber, R.; Brown, G.W.; Green, R. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Pinheiro, P.; Perrais, D.; Coussen, F.; Barhanin, J.; Bettler, B.; Mann, J.R.; Malva, J.; Heinemann, S.F.; Mulle, C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 12181–12186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Ding, M.; Zhang, X.-C.; Liu, Y.; Xuan, J.-F.; Xing, J.-X.; Xia, X.; Yao, J.; Wang, B.-J. Association between polymorphisms in the GRIN1 gene 5′ regulatory region and schizophrenia in a northern Han Chinese population and haplotype effects on protein expression in vitro. BMC Med. Genet. 2019, 20, 26. [Google Scholar] [CrossRef]
- Formicola, D.; Aloia, A.; Sampaolo, S.; Farina, O.; Diodato, D.; Griffiths, L.R.; Gianfrancesco, F.; Di Iorio, G.; Esposito, T. Common variants in the regulative regions of GRIA1 and GRIA3 receptor genes are associated with migraine susceptibility. BMC Med. Genet. 2010, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Domart, M.-C.; Benyamina, A.; Lemoine, A.; Bourgain, C.; Blecha, L.; Debuire, B.; Reynaud, M.; Saffroy, R. Association between a polymorphism in the promoter of a glutamate receptor subunit gene (GRIN2A) and alcoholism. Addict. Biol. 2011, 17, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.E.F.; Kadonaga, J.T. The RNA polymerase II core promoter: A key component in the regulation of gene expression. Genes Dev. 2002, 2583–2592. [Google Scholar] [CrossRef]
- Bai, G.; Hoffman, P.W. Transcriptional Regulation of NMDA Receptor Expression. In Biology of the NMDA Receptor; CRC Press: Boca Raton, FL, USA, 2009; pp. 79–102. [Google Scholar]
- Jiang, Y.; Lin, M.; Jicha, G.A.; Ding, X.; McIlwrath, S.L.; Fardo, D.; Broster, L.S.; Schmitt, F.A.; Kryscio, R.; Lipsky, R.H. Functional human GRIN2B promoter polymorphism and variation of mental processing speed in older adults. Aging 2017, 9, 1293–1306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toulopoulou, T.; Zhang, X.; Cherny, S.; Dickinson, D.; Berman, K.F.; E Straub, R.; Sham, P.; Weinberger, D.R. Polygenic risk score increases schizophrenia liability through cognition-relevant pathways. Brain 2019, 142, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Wimberley, T.; Gasse, C.; Meier, S.M.; Agerbo, E.; MacCabe, J.H.; Horsdal, H.T. Polygenic Risk Score for Schizophrenia and Treatment-Resistant Schizophrenia. Schizophr. Bull. 2017, 43, 1064–1069. [Google Scholar] [CrossRef]
- Chalmer, M.A.; Esserlind, A.-L.; Olesen, J.; Hansen, T.F. Polygenic risk score: Use in migraine research. J. Headache Pain 2018, 19, 29. [Google Scholar] [CrossRef]
- Gogtay, N.; Thompson, P.M. Mapping gray matter development: Implications for typical development and vulnerability to psychopathology. Brain Cogn. 2010, 72, 6–15. [Google Scholar] [CrossRef]
- Schnack, H.G.; van Haren, N.E.M.; Nieuwenhuis, M.; Pol, H.E.H.; Cahn, W.; Kahn, R.S. Accelerated Brain Aging in Schizophrenia: A Longitudinal Pattern Recognition Study. Am. J. Psychiatry 2016, 173, 607–616. [Google Scholar] [CrossRef]
- Hajek, T.; Franke, K.; Kolenic, M.; Capkova, J.; Matejka, M.; Propper, L.; Uher, R.; Stopkova, P.; Novák, T.; Paus, T.; et al. Brain Age in Early Stages of Bipolar Disorders or Schizophrenia. Schizophr. Bull. 2019, 45, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Cahn, W.; Rais, M.; Stigter, F.; van Haren, N.; Caspers, E.; Pol, H.H.; Xu, Z.; Schnack, H.; Kahn, R. Psychosis and brain volume changes during the first five years of schizophrenia. Eur. Neuropsychopharmacol. 2009, 19, 147–151. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Smieskova, R.; Kempton, M.; Ho, B.-C.; Andreasen, N.; Borgwardt, S. Progressive brain changes in schizophrenia related to antipsychotic treatment? A meta-analysis of longitudinal MRI studies. Neurosci. Biobehav. Rev. 2013, 37, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Brans, R.G.H.; Van Haren, N.E.M.; Van Baal, G.C.M.; Schnack, H.G.; Kahn, R.S.; Pol, H.E.H. Heritability of Changes in Brain Volume Over Time in Twin Pairs Discordant for Schizophrenia. Arch. Gen. Psychiatry 2008, 65, 1259–1268. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FES (n = 63) | HC (n = 32) | p-Value (Two-Tailed t-Test) | |
|---|---|---|---|
| Age, years; mean (SD) | 24.62 (6.94) | 27.84 (6.96) | 0.0355 |
| Female, No. (%) | 24 (38.10) | 17.00 (53.13) | 0.16 |
| Male, No. (%) | 39 (61.90) | 15.00 (46.88) | |
| Education, years; mean (SD) | 12.72 (2.84) | 16.74 (2.40) | 0.00 + |
| Schizophrenia, No. (%) | 38 (60.32) | ||
| Other schizophrenia-spectrum disorders *, No. (%) | 25.00 (39,68) | ||
| PANSS Positive Subscale; mean (SD) | 14.82 (4.91) | ||
| PANSS Negative Subscale; mean (SD) | 18.48 (6.46) | ||
| PANSS General Psychopathology Subscale; mean (SD) | 36.17 (8.87) | ||
| PANSS total; mean (SD) | 69.48 (17.35) | ||
| Duration of untreated psychosis (months); mean (SD) | 3.61 (5.05) | ||
| Age at disease onset; mean (SD) | 24.13 (6.97) | ||
| Chlorpromazine equivalents, mg/d; mean (SD) | 399.67 (192.03) | ||
| Duration of antipsychotic treatment (months); mean (SD) | 1.79 (2.96) |
| Sample | Chr | iGluR Gene | RefSeq | Ensembl Canonical | Promotor (Existing/Novel) | Per 1000 bp | 5´UTR (Existing/Novel) | Per 1000 bp | EXONS (Existing/Novel) | Missense/Synonymous (Novel) | Frameshift Variant | Per 1000 bp | INTRONS (Existing/Novel) | Per 1000 bp | 3´UTR (Existing/Novel) | Per 1000 bp |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCH cases | chr5 | GRIA1 | NM_001258022.1 | ENST00000518783.1 | 10/0 | 6.7 | 0/0 | 0.0 | 3/0 | 0/2 | 1 | 1.1 | (1563) 1522/41 | 4.9 | 4/1 | 2.0 |
| Controls | chr5 | GRIA1 | NM_001258022.1 | ENST00000518783.1 | 8/0 | 5.3 | 0/0 | 0.0 | 3/0 | 1/1 | 1 | 1.1 | (1423) 1396/27 | 4.5 | 3/0 | 1.2 |
| SCH cases | chr4 | GRIA2 | NM_000826.4 | ENST00000296526.7 | 1/0 | 0.7 | 0/0 | 0.0 | 3/0 | 1/2 | 0 | 1.1 | (349) 333/16 | 2.5 | 8/1 | 3.4 |
| Controls | chr4 | GRIA2 | NM_000826.4 | ENST00000296526.7 | 2/0 | 1.3 | 1/0 | 3.1 | 2/0 | 0/2 | 0 | 0.8 | (302) 297/5 | 2.2 | 6/0 | 2.3 |
| SCH cases | chrX | GRIA3 | NM_000828.4 | ENST00000622768.4 | 4/0 | 2.7 | 0/0 | 0.0 | 2/0 | 1/1 | 0 | 0.7 | (540) 526/14 | 1.7 | 6/0 | 2.7 |
| Controls | chrX | GRIA3 | NM_000828.4 | ENST00000622768.4 | 2/1 | 2.0 | 0/0 | 0.0 | 1/0 | 0/1 | 0 | 0.4 | (446) 440/6 | 1.5 | 1/0 | 0.5 |
| SCH cases | chr11 | GRIA4 | NM_000829.4 | ENST00000282499.1 | 5/1 | 4.0 | 1/0 | 2.3 | 4/0 | 0/4 | 0 | 1.5 | (1107) 1068/39 | 3.0 | 9/0 | 3.8 |
| Controls | chr11 | GRIA4 | NM_000829.4 | ENST00000282499.1 | 4/0 | 2.7 | 1/0 | 2.3 | 2/0 | 0/2 | 0 | 0.7 | (948) 932/16 | 2.6 | 8/0 | 3.4 |
| SCH cases | chr21 | GRIK1 | NM_001330994.2 | ENST00000327783.4 | 4/0 | 2.7 | 0/0 | 0.0 | 4/0 | 1/3 | 0 | 1.4 | (1620) 1589/31 | 4.1 | 1/0 | 3.4 |
| Controls | chr21 | GRIK1 | NM_001330994.2 | ENST00000327783.4 | 1/0 | 0.7 | 0/0 | 0.0 | 4/0 | 0/4 | 0 | 1.4 | (1339) 1317/22 | 3.4 | 0/1 | 3.4 |
| SCH cases | chr6 | GRIK2 | NM_021956.4 | ENST00000421544.1 | 0/0 | 0.0 | 0/0 | 0.0 | 4/1 | 1/4 (1) | 0 | 1.8 | (2816) 2756/60 | 4.2 | 5/0 | 3.2 |
| Controls | chr6 | GRIK2 | NM_021956.4 | ENST00000421544.1 | 0/0 | 0.0 | 0/0 | 0.0 | 4/0 | 1/3 | 0 | 1.5 | (2339) 2307/32 | 3.5 | 3/0 | 1.9 |
| SCH cases | chr1 | GRIK3 | NM_000831.4 | ENST00000373091.8 | 5/0 | 3.3 | 0/0 | 0.0 | 3/0 | 1/2 | 0 | 1.1 | (938) 912/26 | 4.1 | 13/0 | 2.1 |
| Controls | chr1 | GRIK3 | NM_000831.4 | ENST00000373091.8 | 6/0 | 4.0 | 0/0 | 0.0 | 2/0 | 1/1 | 0 | 0.7 | (602) 592/10 | 2.6 | 4/0 | 0.6 |
| SCH cases | chr11 | GRIK4 | NM_014619.4 | ENST00000527524.2 | 10/1 | 7.3 | 1/0 | 3.5 | 7/0 | 1/6 | 0 | 2.4 | (1976) 1938/38 | 4.2 | 10/0 | 3.8 |
| Controls | chr11 | GRIK4 | NM_014619.4 | ENST00000527524.2 | 6/0 | 4.0 | 1/0 | 3.5 | 6/0 | 1/5 | 0 | 2.1 | (1429) 1397/32 | 3.0 | 8/0 | 3.0 |
| SCH cases | chr19 | GRIK5 | NM_001301030.1 | ENST00000301218.4 | 3/0 | 2.0 | 0/0 | 0.0 | 2/0 | 1/1 | 0 | 0.7 | (65) 63/2 | 1.1 | 1/0 | 3.0 |
| Controls | chr19 | GRIK5 | NM_001301030.1 | ENST00000301218.4 | 3/1 | 2.7 | 0/0 | 0.0 | 2/1 | 2/1 (1) | 0 | 1.0 | (50) 49/1 | 0.8 | 1/0 | 3.0 |
| SCH cases | chr9 | GRIN1 | NM_001185090.2 | ENST00000371553.3 | 2/0 | 1.3 | 0/0 | 0.0 | 3/0 | 0/3 | 0 | 1.1 | (67) 67/0 | 2.6 | 0/0 | 0.0 |
| Controls | chr9 | GRIN1 | NM_001185090.2 | ENST00000371553.3 | 2/0 | 1.3 | 0/0 | 0.0 | 2/0 | 0/2 | 0 | 0.7 | (60) 56/4 | 2.4 | 0/0 | 0.0 |
| SCH cases | chr16 | GRIN2A | NM_001134407.3 | ENST00000330684.3 | 28/0 | 18.7 | 0/0 | 0.0 | 7/1 | (1) 4/4 | 0 | 1.8 | (2162) 2112/50 | 5.2 | 31/0 | 3.2 |
| Controls | chr16 | GRIN2A | NM_001134407.3 | ENST00000330684.3 | 17/0 | 11.3 | 0/0 | 0.0 | 6/0 | 4/2 | 0 | 1.4 | (1842) 1816/26 | 4.5 | 29/0 | 3.0 |
| SCH cases | chr12 | GRIN2B | NM_000834.4 | ENST00000609686.1 | 4/0 | 2.7 | 2/2 | 19.1 | 12/1 | 0/13 (1) | 0 | 2.9 | (1779) 1730/49 | 4.3 | 134/1 | 6.0 |
| Controls | chr12 | GRIN2B | NM_000834.4 | ENST00000609686.1 | 4/0 | 2.7 | 2/0 | 9.5 | 10/0 | 0/10 | 0 | 2.2 | (1449) 1430/19 | 3.5 | 107/1 | 4.8 |
| SCH cases | chr17 | GRIN2C | NM_000835.5 | ENST00000293190.5 | 1/0 | 0.7 | 1/0 | 6.8 | 6/0 | 3/2 | 1 | 1.6 | (31) 30/1 | 2.3 | 0/0 | 0.0 |
| Controls | chr17 | GRIN2C | NM_000835.5 | ENST00000293190.5 | 1/1 | 1.3 | 0/0 | 0.0 | 7/0 | 3/4 | 0 | 1.9 | (27) 27/0 | 2.0 | 1/0 | 2.4 |
| SCH cases | chr19 | GRIN2D | NM_000836.2 | ENST00000263269.3 | 5/0 | 3.3 | 1/0 | 11.4 | 3/0 | 0/3 | 0 | 0.8 | (109) 107/2 | 2.4 | 1/0 | 1.0 |
| Controls | chr19 | GRIN2D | NM_000836.2 | ENST00000263269.3 | 2/0 | 1.3 | 1/0 | 11.4 | 3/0 | 1/2 | 0 | 0.8 | (87) 84/3 | 1.9 | 1/0 | 1.0 |
| SCH cases | chr9 | GRIN3A | NM_133445.2 | ENST00000361820.3 | 6/0 | 4.0 | 6/0 | 10.0 | 18/0 | 8/10 | 0 | 5.4 | (760) 752/8 | 4.7 | 14/1 | 3.9 |
| Controls | chr9 | GRIN3A | NM_133445.2 | ENST00000361820.3 | 4/0 | 2.7 | 5/0 | 8.3 | 15/0 | 6/9 | 0 | 4.5 | (603) 597/6 | 3.7 | 13/2 | 3.9 |
| SCH cases | chr19 | GRIN3B | NM_138690.3 | ENST00000234389.3 | 13/2 | 10.0 | 0/0 | 0.0 | 25/1 | 16/9 (1) | 1 | 8.3 | (38) 36/2 | 6.3 | 0/0 | 0.0 |
| Controls | chr19 | GRIN3B | NM_138690.3 | ENST00000234389.3 | 11/0 | 7.3 | 0/0 | 0.0 | 19/0 | 10/9 | 0 | 6.1 | (29) 29/0 | 4.8 | 0/0 | 0.0 |
| ID | hg38 | Transcript (Ensembl) | Consequence | iGluR Gene | Exon | Ref. Allele | ALT Allele | SCH Cases, MAF | Controls, MAF | p-Value * |
|---|---|---|---|---|---|---|---|---|---|---|
| rs145573036 | chr4:157336626 | ENST00000296526.12 | p.Glu575Gln | GRIA2 | 11/16 | G | C | 0.0079 | 0.0000 | ND |
| rs3841128 | chr5:153492228 | ENST00000518783.1 | p.Leu11ProfsTer13 | GRIA1 | 1/16 | – | C | 0.0635 | 0.0781 | 0.7643 |
| rs146865938 | chr5:153650491 | ENST00000518783.1 | p.Arg218Cys | GRIA1 | 4/16 | C | T | 0.0000 | 0.0156 | 0.3368 |
| rs71509734 | chr9:101573337 | ENST00000361820.6 | p.Asn1062Ser | GRIN3A | 9/9 | T | C | 0.0079 | 0.0000 | ND |
| chr9:101737814 | ENST00000361820.6 | p.Gln56 * | GRIN3A | 1/9 | G | A | 0.0079 | 0.0000 | ND | |
| rs34755188 | chr9:101670973 | ENST00000361820.6 | p.Arg480His | GRIN3A | 3/9 | C | T | 0.0079 | 0.0313 | 0.2628 |
| rs10989589 | chr9:101670953 | ENST00000361820.6 | p.Gly487Arg | GRIN3A | 3/9 | C | T | 0.4048 | 0.4844 | 0.3528 |
| rs41297895 | chr11:120819909 | ENST00000527524.8 | p.Ala167Gly | GRIK4 | 6/21 | C | G | 0.0000 | 0.0156 | 0.3368 |
| rs137906208 | chr11:120861999 | ENST00000527524.8 | p.Arg262His | GRIK4 | 9/21 | G | A | 0.0079 | 0.0000 | ND |
| rs765016248 | chr17:74843525 | ENST00000293190.10 | p.Gln871Arg | GRIN2C | 13/13 | T | C | 0.0159 | 0.0000 | 0.5509 |
| rs754674133 | chr17:74844496–74844498 | ENST00000293190.10 | p.Lys788ThrfsTer13 | GRIN2C | 12/13 | TC | – | 0.0079 | 0.0000 | ND |
| rs375174698 | chr17:74850716 | ENST00000293190.10 | p.Arg389Cys | GRIN2C | 5/13 | G | A | 0.0000 | 0.0156 | 0.3368 |
| rs545736648 # | chr19:1007862 | ENST00000234389.3 | p.Lys738GlnfsTer34 | GRIN3B | 5/9 | – | C | 0.0246 | 0.0161 | 0.7099 |
| rs201638380 | chr19:1004804 | ENST00000234389.3 | p.Glu435Lys | GRIN3B | 3/9 | G | A | 0.0079 | 0.0000 | ND |
| rs10666583 | chr19:1004897 | ENST00000234389.3 | p.Gly466AlafsTer18 | GRIN3B | 3/9 | – | CGTT | 0.3016 | 0.3125 | 0.8694 |
| rs566603277 | chr19:1005200 | ENST00000234389.3 | p.Gly567Ser | GRIN3B | 3/9 | G | A | 0.0079 | 0.0000 | ND |
| rs765625485 | chr19:1005375 | ENST00000234389.3 | p.Ala625Val | GRIN3B | 3/9 | C | T | 0.0000 | 0.0156 | 0.3368 |
| rs2285906 | chr19:1008684 | ENST00000234389.3 | p.Ala845Thr | GRIN3B | 7/9 | G | A | 0.2063 | 0.0938 | 0.0643 |
| rs61744375 | chr19:1008705 | ENST00000234389.3 | p.Glu852Lys | GRIN3B | 7/9 | G | A | 0.0079 | 0.0000 | ND |
| rs137867437 | chr19:42062790 | ENST00000301218.8 | p.Ser104Pro | GRIK5 | 3/19 | A | G | 0.0000 | 0.0156 | 0.3368 |
| iGluR Gene | Haplotype | hg38 | rs | Location | Allele | Fst | SCH Cases freq | Controls freq | p-Value 1 | OR | Genotype | LR | Genotype | LR | Genotype | LR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GRIK3 | Hap1 | chr1:37014543 | rs3753776 &,§ | intron 1 | T | 0.071281 | 0.381 | 0.188 | 0.0080 | 2.67 | wt/wt | 0.554 | wt/Hap1 | 1.904 | Hap1/Hap1 | 2.270 |
| chr1:37017453 | rs2359647 | intron 1 | T | 0.071281 | ||||||||||||
| chr1:37023779 | rs6686296 # | intron 1 | G | 0.071281 | ||||||||||||
| chr1:37028109 | rs59729868 £ | intron 1 | A | 0.071281 | ||||||||||||
| chr1:37031920 | rs3845491 | intron 1 | T | 0.083134 | ||||||||||||
| chr1:37035122 | rs2134846 | promoter | A | 0.083134 | ||||||||||||
| GRIA1 | Hap2 | chr5:153527505 | rs566577 ¥,£ | intron 2 | C | 0.062782 | 0.183 | 0.047 | 0.0126 | 4.54 | wt/wt | 0.754 | wt/Hap2 | 2.872 | Hap2/Hap2 | ND |
| chr5:153532297 | rs1493395 | intron 2 | G | 0.062782 | ||||||||||||
| chr5:153534826 | rs1908100 | intron 2 | T | 0.062782 | ||||||||||||
| GRIK2 | chr6:101460819 | rs2518230 | intron 4 | T | 0.055046 | 0.548 | 0.344 | 0.0091 | 2.31 | wt/wt | 0.473 | wt/Hap3 | 1.227 | Hap3/Hap3 | 2.032 | |
| Hap3 | chr6:101462357 | rs2245037 & | intron 4 | G | 0.055046 | |||||||||||
| chr6:101467933 | rs2579937 | intron 4 | A | 0.056160 | ||||||||||||
| GRIN3A | Hap4 | chr9:101711604 | rs2485530 #,§,¥,£ | intron 2 | C | 0.050761 | 0.079 | 0.000 | 0.0174 | ND | wt/wt | 0.841 | wt/Hap4 | ND | Hap4/Hap4 | ND |
| chr9:101714448 | rs2506363 | intron 1 | C | 0.050761 | ||||||||||||
| chr9:101715378 | rs2210991 | intron 1 | C | 0.055466 | ||||||||||||
| chr9:101716352 | rs2506364 | intron 1 | A | 0.050761 | ||||||||||||
| chr9:101717714 | rs2065965 | intron 1 | G | 0.050761 | ||||||||||||
| chr9:101720595 | rs7849059 | intron 1 | A | 0.050761 | ||||||||||||
| GRIK4 | Hap5 | chr11:120746795 | rs1945010 ¥,£ | intron 3 | C | 0.058187 | 0.683 | 0.500 | 0.0177 | 2.15 | wt/wt | 0.361 | wt/Hap5 | 0.845 | Hap5/Hap5 | 2.027 |
| chr11:120747370 | rs1939673 #,§,¥,£ | intron 3 | T | 0.051846 | ||||||||||||
| GRIK4 | Hap6 | chr11:120927187 | rs5795249 | intron13 | T | 0.101537 | 0.222 | 0.047 | 0.0016 | 5.81 | wt/wt | 0.683 | wt/Hap6 | 3.372 | Hap6/Hap6 | ND |
| chr11:120927920 | rs1893816 | intron13 | T | 0.101537 | ||||||||||||
| chr11:120930476 | rs2186620 ¥,£ | intron13 | C | 0.094634 | ||||||||||||
| GRIN2B | none | chr12:13634912 | rs2160519 | intron 4 | C | 0.057178 | 0.087 (C) | 0.000 (C) | 0.0171 | ND | T/T | 0.825 | T/C | ND | C/C | ND |
| GRIN2B | none | chr12:13722646 | rs2110984 | intron 3 | T | 0.080249 | 0.825 (T) | 0.641 (T) | 0.0066 | 2.65 | C/C | 0.508 | C/T | 0.481 | T/T | 1.985 |
| GRIN2B | chr12:13728086 | rs2284412 #,¥,£ | intron 3 | T | 0.054135 | 0.317 | 0.156 | 0.0228 | 2.51 | wt/wt | 0.669 | wt/Hap7 | 1.419 | Hap7/Hap7 | ND | |
| Hap7 | chr12:13741902 | rs10772710 | intron 3 | A | 0.054135 | |||||||||||
| chr12:13744486 | rs2268121 $,¥,£ | intron 3 | C | 0.054135 | ||||||||||||
| GRIN2B | none | chr12:13871835 | rs10845851 | intron 1 | G | 0.061579 | 0.095 (G) | 0.000 (G) | 0.0072 | ND | A/A | 0.825 | A/G | ND | G/G | ND |
| GRIN2D | none | chr19:48437650 | rs56125279 | intron 10 | G | 0.051118 | 0.190 (G) | 0.063 (G) | 0.0181 | 3.53 | C/C | 0.762 | C/G | 2.288 | G/G | ND |
| GRIK1 | Hap8 | chr21:29925689 | rs2832486 | intron 1 | T | 0.052022 | 0.730 | 0.563 | 0.0227 | 2.10 | wt/wt | 0.308 | wt/Hap8 | 0.789 | Hap8/Hap8 | 1.808 |
| chr21:29926886 | rs2832488 | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29927152 | rs2832489 | intron 1 | A | 0.052022 | ||||||||||||
| chr21:29929318 | rs2832490 | intron 1 | G | 0.052022 | ||||||||||||
| chr21:29930154 | rs2255821 | intron 1 | A | 0.052022 | ||||||||||||
| chr21:29930365 | rs767253 ¥,£ | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29931426 | rs2300333 | intron 1 | G | 0.052022 | ||||||||||||
| chr21:29931851 | rs2832492 | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29933351 | rs9982938 ¥,£ | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29933357 | rs9982699 ¥,£ | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29933999 | rs3831792 | intron 1 | AATATAAATG | 0.052022 | ||||||||||||
| chr21:29934461 | rs2243533 | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29934499 | rs2243535 | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29934740 | rs2243435 | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29934751 | rs2243436 | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29936109 | rs2832494 | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29937434 | rs2832495 | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29937912 | rs2409355 | intron 1 | C | 0.052022 | ||||||||||||
| chr21:29938441 | rs2832496 | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29938848-29938850 | rs66799775 | intron 1 | G | 0.052022 | ||||||||||||
| chr21:29938853 | rs75584618 & | intron 1 | T | 0.052022 | ||||||||||||
| chr21:29938854 | rs77191337 | intron 1 | T | 0.052022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirschfeldova, K.; Cerny, J.; Bozikova, P.; Kuchtiak, V.; Rausch, T.; Benes, V.; Spaniel, F.; Gregus, D.; Horacek, J.; Vyklicky, L.; et al. Evidence for the Association between the Intronic Haplotypes of Ionotropic Glutamate Receptors and First-Episode Schizophrenia. J. Pers. Med. 2021, 11, 1250. https://doi.org/10.3390/jpm11121250
Hirschfeldova K, Cerny J, Bozikova P, Kuchtiak V, Rausch T, Benes V, Spaniel F, Gregus D, Horacek J, Vyklicky L, et al. Evidence for the Association between the Intronic Haplotypes of Ionotropic Glutamate Receptors and First-Episode Schizophrenia. Journal of Personalized Medicine. 2021; 11(12):1250. https://doi.org/10.3390/jpm11121250
Chicago/Turabian StyleHirschfeldova, Katerina, Jiri Cerny, Paulina Bozikova, Viktor Kuchtiak, Tobias Rausch, Vladimir Benes, Filip Spaniel, David Gregus, Jiri Horacek, Ladislav Vyklicky, and et al. 2021. "Evidence for the Association between the Intronic Haplotypes of Ionotropic Glutamate Receptors and First-Episode Schizophrenia" Journal of Personalized Medicine 11, no. 12: 1250. https://doi.org/10.3390/jpm11121250
APA StyleHirschfeldova, K., Cerny, J., Bozikova, P., Kuchtiak, V., Rausch, T., Benes, V., Spaniel, F., Gregus, D., Horacek, J., Vyklicky, L., & Balik, A. (2021). Evidence for the Association between the Intronic Haplotypes of Ionotropic Glutamate Receptors and First-Episode Schizophrenia. Journal of Personalized Medicine, 11(12), 1250. https://doi.org/10.3390/jpm11121250