
Gut Microbiome Profiling Uncovers a Lower Abundance of Butyricicoccus in Advanced Stages of Chronic Kidney Disease


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. Determination of Uremic Metabolites in Blood
2.3. Illumina-Based Microbial Profiling
2.4. Statistical Analysis
3. Results
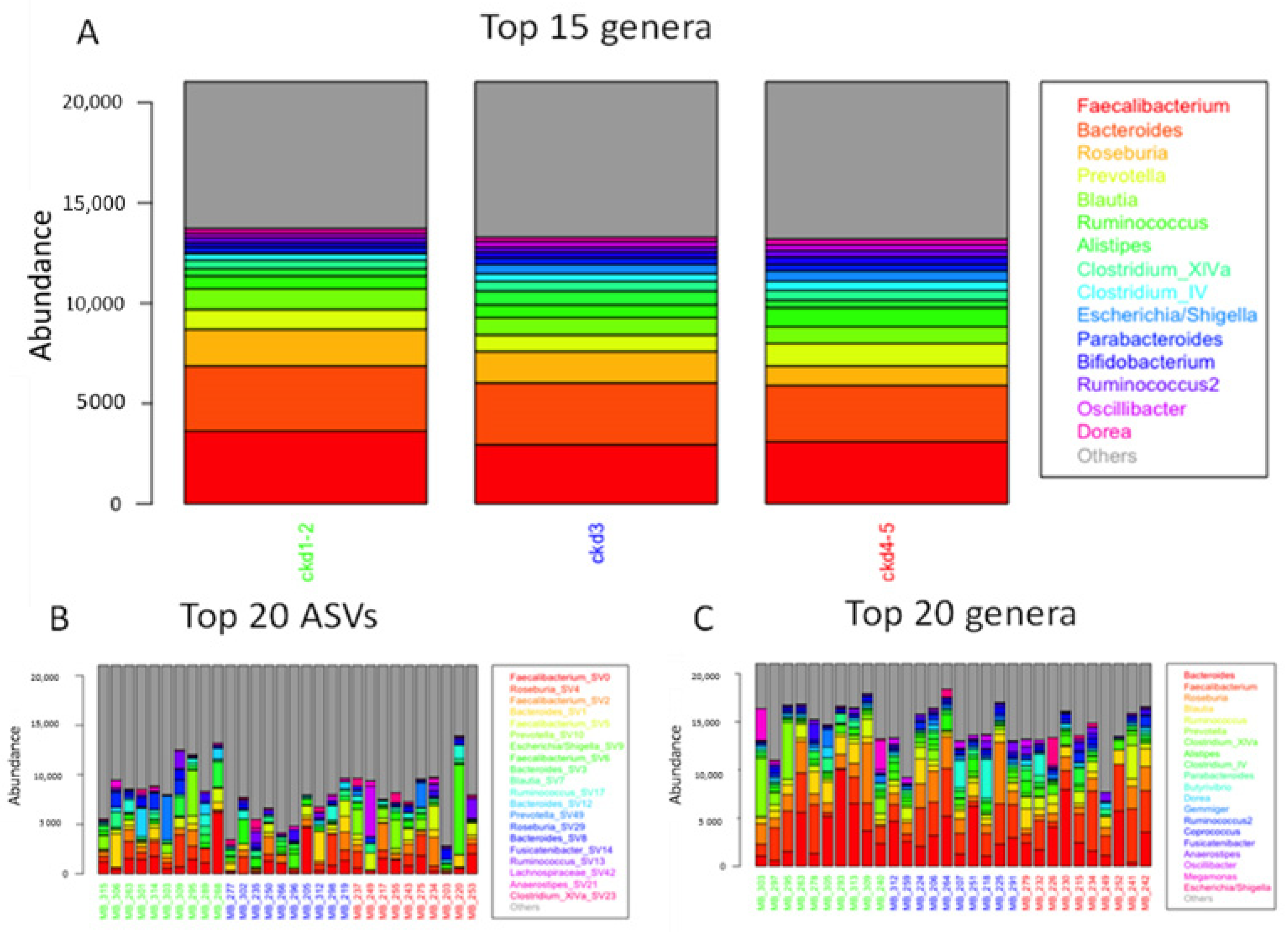
3.1. Gut Microbiome Profiles in Different Stages of CKD
3.2. Correlates of Intestinal Microbiota Composition in CKD
3.3. Variation in Gut Microbiota Profiles in Different Stages of CKD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Glorieux, G.; De Smet, R.; Lameire, N. New insights in uremic toxins. Kidney Int. Suppl. 2003, 63 (Suppl. S84), S6–S10. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Chuang, C.K.; Jayakumar, T.; Liu, H.L.; Pan, C.F.; Wang, T.J.; Chen, H.H.; Wu, C.J. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch. Med. Sci. 2013, 9, 662–668. [Google Scholar] [CrossRef]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76 (Suppl. S114), S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of Colon-Derived Uremic Solutes. Clin. J. Am. Soc. Nephrol. 2018, 13, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Loor, H.; Bammens, B.; Evenepoel, P.; De Preter, V.; Verbeke, K. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin. Chem. 2005, 51, 1535–1538. [Google Scholar] [CrossRef]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Deltombe, O.; Van Biesen, W.; Glorieux, G.; Massy, Z.; Dhondt, A.; Eloot, S. Exploring Protein Binding of Uremic Toxins in Patients with Different Stages of Chronic Kidney Disease and during Hemodialysis. Toxins 2015, 7, 3933–3946. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Kuo, C.C.; Chen, Y.M.; Hsu, C.Y.; Lee, W.C.; Tsai, Y.C.; Ng, H.Y.; Kuo, L.C.; Chiou, T.T.; Yang, Y.K.; et al. Factors associated with blood concentrations of indoxyl sulfate and p-cresol in patients undergoing peritoneal dialysis. Perit. Dial. Int. 2010, 30, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuuchi, F.H.M.; Aiba, Y.; Koga, Y.; Endoh, M.; Kurokawa, K.; Sakai, H. Intestinal bacteria-derived putrefactants in chronic renal failure. Clin. Exp. Nephrol. 2002, 6, 6. [Google Scholar] [CrossRef]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen 2019, 8, e00678. [Google Scholar] [CrossRef]
- Shi, K.; Wang, F.; Jiang, H.; Liu, H.; Wei, M.; Wang, Z.; Xie, L. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 2014, 59, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef]
- Joossens, M.; Faust, K.; Gryp, T.; Nguyen, A.T.L.; Wang, J.; Eloot, S.; Schepers, E.; Dhondt, A.; Pletinck, A.; Vieira-Silva, S.; et al. Gut microbiota dynamics and uraemic toxins: One size does not fit all. Gut 2019, 68, 2257–2260. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Lewis, S.J.; Heaton, K.W. Stool form scale as a useful guide to intestinal transit time. Scand. J. Gastroenterol. 1997, 32, 920–924. [Google Scholar] [CrossRef]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Snauwaert, E.; Holvoet, E.; Van Biesen, W.; Raes, A.; Glorieux, G.; Vande Walle, J.; Roels, S.; Vanholder, R.; Askiti, V.; Azukaitis, K.; et al. Uremic Toxin Concentrations are Related to Residual Kidney Function in the Pediatric Hemodialysis Population. Toxins 2019, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.G.F.; Friendly, B.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 27 October 2021).
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Leeuwenhoek 2016, 109, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Kakuta, M.; Hasegawa, T.; Yamaguchi, R.; Uchino, E.; Murashita, K.; Nakaji, S.; Imoto, S.; Yanagita, M.; Okuno, Y. Metagenomic profiling of gut microbiome in early chronic kidney disease. Nephrol. Dial. Transpl. 2020, 36, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadlbauer, V.; Horvath, A.; Ribitsch, W.; Schmerböck, B.; Schilcher, G.; Lemesch, S.; Stiegler, P.; Rosenkranz, A.R.; Fickert, P.; Leber, B. Structural and functional differences in gut microbiome composition in patients undergoing haemodialysis or peritoneal dialysis. Sci. Rep. 2017, 7, 15601. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; Dore, J.; Leclerc, M.; McSweeney, C.S. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm. Bowel Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Van Immerseel, F.; Ducatelle, R.; De Vos, M.; Boon, N.; Van De Wiele, T.; Verbeke, K.; Rutgeerts, P.; Sas, B.; Louis, P.; Flint, H.J. Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. J. Med. Microbiol. 2010, 59, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Pereira, N.B.F.; Ramos, C.I.; de Andrade, L.S.; Teixeira, R.R.; Nakao, L.S.; Rodrigues, S.D.; de Lima, J.D.; Dalboni, M.A.; Cuppari, L. Influence of bowel habits on gut-derived toxins in peritoneal dialysis patients. J. Nephrol. 2020, 33, 1049–1057. [Google Scholar] [CrossRef]
- Ramos, C.I.; Armani, R.G.; Canziani, M.E.; Ribeiro Dolenga, C.J.; Nakao, L.S.; Campbell, K.L.; Cuppari, L. Bowel Habits and the Association with Uremic Toxins in Non-Dialysis-Dependent Chronic Kidney Disease Patients. J. Ren. Nutr. 2020, 30, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518–1524. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Correlation to BSS | All CKD Stages (n = 111) | CKD Stage 1–2 (n = 37) | CKD Stage 3 (n = 44) | CKD Stage 4–5 (n = 33) | ||||
|---|---|---|---|---|---|---|---|---|
| Uremic Toxin | rs | p-Value | rs | p-Value | rs | p-Value | rs | p-Value |
| Indoxyl sulfate | −0.173 | ns | −0.130 | ns | −0.054 | ns | −0.223 | ns |
| Indole-3-acetic acid | −0.150 | ns | −0.035 | ns | −0.041 | ns | −0.206 | ns |
| Hippuric acid | −0.343 | <0.001 | −0.366 | 0.036 | −0.153 | ns | −0.318 | ns |
| p-Cresyl sulfate | −0.287 | 0.003 | −0.335 | ns | −0.134 | ns | −0.443 | 0.012 |
| p-Cresyl glucuronide | −0.175 | ns | −0.105 | ns | −0.116 | ns | −0.340 | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gryp, T.; Faust, K.; Van Biesen, W.; Huys, G.R.B.; Verbeke, F.; Speeckaert, M.; Raes, J.; Vaneechoutte, M.; Joossens, M.; Glorieux, G. Gut Microbiome Profiling Uncovers a Lower Abundance of Butyricicoccus in Advanced Stages of Chronic Kidney Disease. J. Pers. Med. 2021, 11, 1118. https://doi.org/10.3390/jpm11111118
Gryp T, Faust K, Van Biesen W, Huys GRB, Verbeke F, Speeckaert M, Raes J, Vaneechoutte M, Joossens M, Glorieux G. Gut Microbiome Profiling Uncovers a Lower Abundance of Butyricicoccus in Advanced Stages of Chronic Kidney Disease. Journal of Personalized Medicine. 2021; 11(11):1118. https://doi.org/10.3390/jpm11111118
Chicago/Turabian StyleGryp, Tessa, Karoline Faust, Wim Van Biesen, Geert R. B. Huys, Francis Verbeke, Marijn Speeckaert, Jeroen Raes, Mario Vaneechoutte, Marie Joossens, and Griet Glorieux. 2021. "Gut Microbiome Profiling Uncovers a Lower Abundance of Butyricicoccus in Advanced Stages of Chronic Kidney Disease" Journal of Personalized Medicine 11, no. 11: 1118. https://doi.org/10.3390/jpm11111118