New Insights into the Molecular Bases of Familial Alzheimer’s Disease



Abstract
1. Introduction
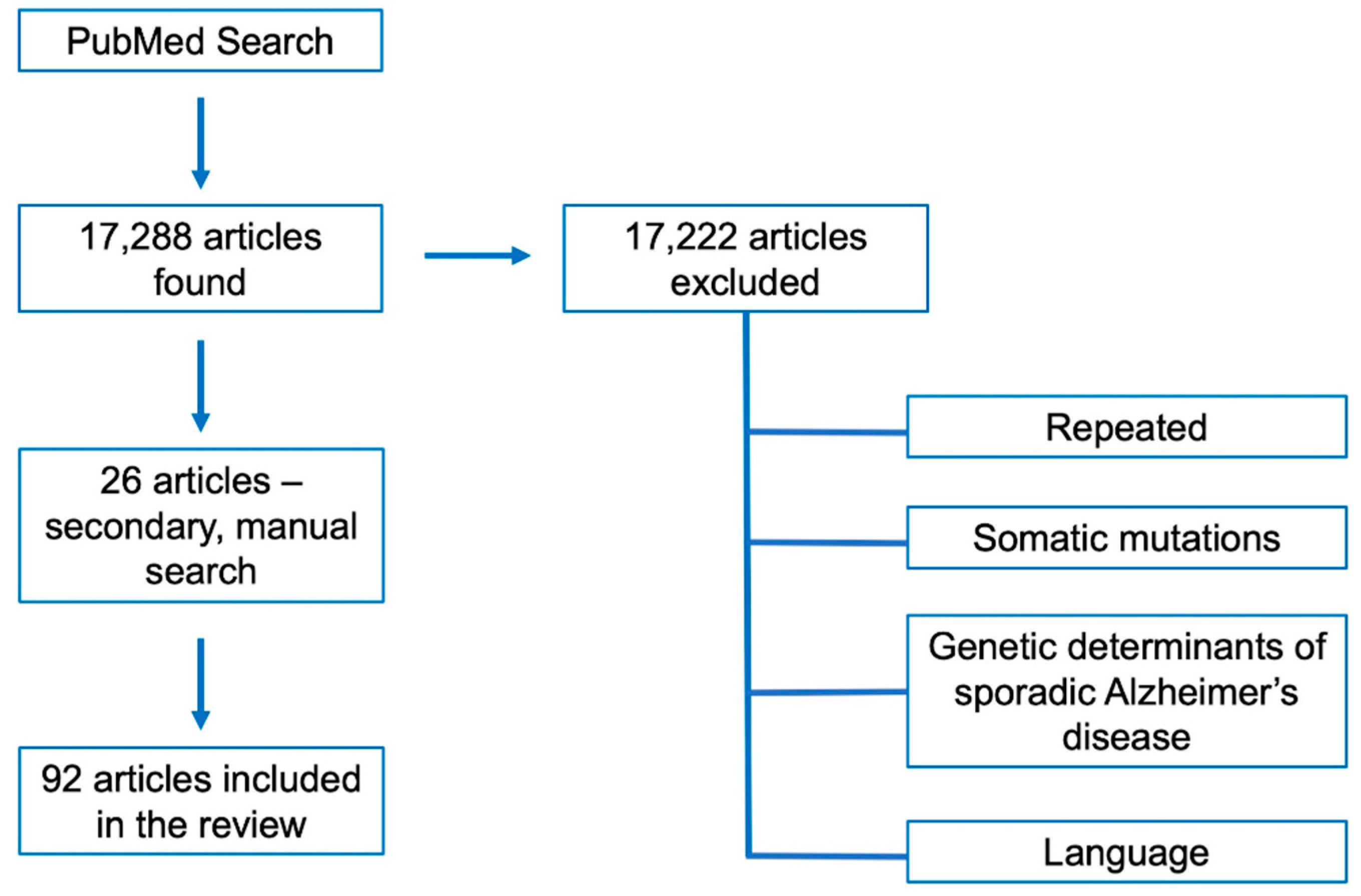
2. Methods
3. Highly Penetrant Familial Alzheimer Disease-Causing Genes
4. Apolipoprotein E ε4 Risk Allele and Familial Alzheimer Disease
5. Novel, Emerging and Candidate Genes Associated to Familial Alzheimer Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collaborators GBDD. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 181, 88–106. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Gustafson, D.R.; Hardy, J. The genetic architecture of Alzheimer’s disease: Beyond APP, PSENs and APOE. Neurobiol. Aging 2012, 33, 437–456. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef]
- Goldman, J.S.; Hahn, S.E.; Catania, J.W.; LaRusse-Eckert, S.; Butson, M.B.; Rumbaugh, M.; Strecker, M.N.; Roberts, J.S.; Burke, W.; Mayeux, R.; et al. American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counseling and testing for Alzheimer disease: Joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet. Med. 2011, 136, 597–605. [Google Scholar] [CrossRef]
- Reiman, E.M.; Langbaum, J.B.; Tariot, P.N.; Lopera, F.; Bateman, R.J.; Morris, J.C.; Sperling, R.A.; Aisen, P.S.; Roses, A.D.; Welsh-Bohmer, K.A.; et al. CAP-advancing the evaluation of preclinical Alzheimer disease treatments. Nat. Rev. Neurol. 2016, 121, 56–61. [Google Scholar] [CrossRef]
- Tariot, P.N.; Lopera, F.; Langbaum, J.B.; Thomas, R.G.; Hendrix, S.; Schneider, L.S.; Rios-Romenets, S.; Giraldo, M.; Acosta, N.; Tobon, C.; et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. (N. Y.) 2018, 4, 150–160. [Google Scholar]
- Bergougnoux, A.; D’Argenio, V.; Sollfrank, S.; Verneau, F.; Telese, A.; Postiglione, I.; Lackner, K.J.; Claustres, M.; Castaldo, G.; Rossman, H.; et al. Multicenter validation study for the certification of a CFTR gene scanning method using next generation sequencing technology. Clin. Chem. Lab. Med. 2018, 56, 1046–1053. [Google Scholar] [CrossRef]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef]
- D’Argenio, V.; Frisso, G.; Precone, V.; Boccia, A.; Fienga, A.; Pacileo, G.; Limongelli, G.; Paolella, G.; Calabrò, R.; Salvatore, F. DNA sequence capture and next-generation sequencing for the molecular diagnosis of genetic cardiomyopathies. J. Mol. Diagn. 2014, 16, 32–44. [Google Scholar] [CrossRef]
- Cariati, F.; D’Argenio, V.; Tomaiuolo, R. The evolving role of genetic tests in reproductive medicine. J. Transl. Med. 2019, 17, 267. [Google Scholar] [CrossRef]
- Nunziato, M.; Esposito, M.V.; Starnone, F.; Diroma, M.A.; Calabrese, A.; Del Monaco, V.; Buono, P.; Frasci, G.; Botti, G.; D’Aiuto, M.; et al. A multi-gene panel beyond BRCA1/BRCA2 to identify new breast cancer-predisposing mutations by a picodroplet PCR followed by a next-generation sequencing strategy: A pilot study. Anal. Chim. Acta 2019, 1046, 154–162. [Google Scholar] [CrossRef]
- Precone, V.; Del Monaco, V.; Esposito, M.V.; De Palma, F.D.; Ruocco, A.; Salvatore, F.; D’Argenio, V. Cracking the code of human diseases using next-generation sequencing: Applications, challenges, and perspectives. Biomed. Res. Int. 2015, 161648. [Google Scholar] [CrossRef]
- D’Argenio, V. The high-throughput analyses era: are we ready for the data struggle? High. Throughput 2018, 7, 8. [Google Scholar] [CrossRef]
- Belinova, I.; Karran, E.; De Strooper, B. The toxic Ab oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. [Google Scholar]
- Sarnataro, D. Attempt to untangle the prion-like misfolding mechanism for neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef]
- De Jonghe, C.; Esselens, C.; Kumar-Singh, S.; Craessaerts, K.; Serneela, S.; Checler, F.; Annaert, W.; Van Broeckhoven, C.; De Strooper, B. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Gen. 2001, 16, 1665–1671. [Google Scholar] [CrossRef]
- Murakami, K.; Irie, K.; Morimoto, A.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Synthesis, aggregation, neurotoxicity, and secondary structure of various Aβ1-42 mutants of familial Alzheimer’s disease at positions 21–23. Biochem. Biophys. Res. Commun. 2002, 294, 5–10. [Google Scholar] [CrossRef]
- Kumar-Singh, S. Hereditary and sporadic forms of Aβ-cerebrovascular amyloidosis and relevant transgenic mouse models. Int. J. Mol. Sci. 2009, 10, 1872–1895. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and processing of the Amyloid-β protein precursor in mitochondria-associated membranes. J. Alzheimers Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Limone, A.; Napolitano, F.; Cerchia, C.; Parisi, S.; Minopoli, G.; Montuori, N.; Lavecchia, A.; Sarnataro, D. APP maturation and intracellular localization are controlled by a specific inhibitor of 37/67 kDa laminin-1 receptor in neuronal cells. Int. J. Mol. Sci. 2020, 21, 1738. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.; Hogel, P.; Silber, M.; Chaitoglou, I.; Luy, B.; Muhle-Goll, C.; Scharnagl, C.; Langosch, D. Increased H-bond stability relates to altered e-cleavage efficiency and Ab levels in the I45T familial Alzheimer’s disease mutant of APP. Sci. Rep. 2019, 9, 5321. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Nadezhdin, K.D.; Urban, A.S.; Volynsky, P.E.; Pavlov, K.V.; Efremov, R.G.; Arseniev, A.S.; Bocharova, O.V. Familial L723P mutation can shift the distribution between the alternative APP transmembrane domain cleavage cascades by local unfolding of the E-cleavage site suggesting a straightforward mechanism of Alzheimer’s disease pathogenesis. ACS Chem. Biol. 2019, 4, 1573–1582. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Rogers, J.T.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of neuronal iron homeostasis as an alternative unifying effect of mutations causing familial Alzheimer’s disease. Front. Neurosci. 2018, 12, 533. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’stype dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Conidi, M.E.; Bernardi, L.; Puccio, G.; Smirne, N.; Muraca, M.G.; Curcio, S.A.; Colao, R.; Piscopo, P.; Gallo, M.; Anfossi, M.; et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology 2015, 84, 2266–2273. [Google Scholar] [CrossRef]
- Cabrejo, L.; Guyant-Marechal, L.; Laquerriere, A.; Vercelletto, M.; De la Fourniere, F.; Thomas-Anterion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerriere, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Remes, A.M.; Finnila, S.; Mononen, H.; Tuominen, H.; Takalo, R.; Herva, R.; Majamaa, K. Hereditary dementia with intracerebral hemorrhages and cerebral amyloid angiopathy. Neurology 2004, 63, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Frebourg, T.; Tuominen, H.; Majamaa, K.; Campion, D.; Remes, A.M. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; Del-Favero, J.; Cruts, M.; van Duijn, C.M.; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006, 129, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Hooli, B.V.; Mohapatra, G.; Mattheisen, M.; Parrado, A.R.; Roehr, J.T.; Shen, Y.; Gusella, J.F.; Moir, R.; Saunders, A.J.; Lange, C.; et al. Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology 2012, 78, 1250–1257. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef]
- Pilotto, A.; Padovani, A.; Borroni, B. Clinical, biological, and imaging features of monogenic Alzheimer’s Disease. Biomed. Res. Int. 2013, 689591. [Google Scholar] [CrossRef]
- Portet, F.; Dauvilliers, Y.; Campion, D.; Raux, G.; Hauw, J.J.; Lyon-Caen, O.; Camu, W.; Touchon, J. Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology 2003, 61, 1136–1137. [Google Scholar] [CrossRef]
- Golan, M.P.; Styczynska, M.; Jozwiak, K.; Walecki, J.; Maruszak, A.; Pniewski, J.; Lugiewicz, R.; Filipek, S.; Zekanowski, C.; Barcikowska, M. Early-onset Alzheimer’s disease with a de novo mutation in the presenilin 1 gene. Exp. Neurol. 2007, 208, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselèe, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. Collaborators of the CNR-MAJ project. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Munoz, C.; Lopez, L.; Arcila, M.L.; Garcia, G.; Madrigal, L.; Moreno, S.; Ríos Romenets, S.; Lopez, H.; Gutierrez, M.; et al. Homozygosity of the autosomal dominant Alzheimer disease presenilin 1 E280A mutation. Neurology 2015, 84, 206–208. [Google Scholar] [CrossRef]
- Hardy, J.; Crook, R. Presenilin mutations line up along transmembrane alpha-helices. Neurosci. Lett. 2001, 306, 203–205. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Beck, J.; Gibbs, J.R.; Santana, I.; Rossor, M.N.; Schott, J.M.; Nalls, M.A.; Ribeiro, H.; Santiago, B.; Fox, N.C.; et al. Genetic Variability in CLU and Its Association with Alzheimer’s Disease. PLoS ONE 2010, 5, e9510. [Google Scholar] [CrossRef]
- Brouwers, N.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of Alzheimer’s disease: An update. Ann. Med. 2008, 40, 562–583. [Google Scholar] [CrossRef]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef]
- Jarmolowicz, A.I.; Chen, H.Y.; Panegyres, P.K. The Patterns of Inheritance in Early-Onset Dementia: Alzheimer’s Disease and Frontotemporal Dementia. Am. J. Alzheimers Dis. Other Dement. 2014, 30, 299–306. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu Rev. Genomics Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Ashford, J.W. APOE genotype effects on Alzheimer’s disease onset and epidemiology. J. Mol. Neurosci. 2004, 23, 157–165. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Huang, Y. Apolipoprotein E and Alzheimer disease. Neurology 2006, 66, S79–S85. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, C.M.; de Knijff, P.; Cruts, M.; Wehnert, A.; Havekes, L.M.; Hofman, A.; Van Broeckhoven, C. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat. Genet. 1994, 7, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Nacmias, B.; Forleo, P.; Piacentini, S.; Latorraca, S.; Amaducci, L. Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer’s disease. Ann. Neurol. 1995, 38, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Pastor, P.; Roe, C.M.; Villegas, A.; Bedoya, G.; Chakraverty, S.; Garcia, G.; Bedoya, G.; Chakraverty, S.; García, G.; Tirado, V.; et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann. Neurol. 2003, 54, 163–169. [Google Scholar] [CrossRef]
- Wijsman, E.M.; Daw, E.W.; Yu, X.; Steinbart, E.J.; Nochlin, D.; Bird, T.D.; Schellenberg, G.D. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 132, 14–20. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild Cognitive Impairment. Continuum (Minneap Minn) 2016, 22, 404–418. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Kinsella, E.; Bras, J.M.; Luu, N.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Santana, I.; Hanagasi, H.; et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1008–1023. [Google Scholar] [CrossRef]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C.; et al. Alzheimer’s Disease Sequencing Project. Association of Rare Coding Mutations with Alzheimer Disease and Other Dementias Among Adults of European Ancestry. JAMA Netw. Open 2019, 2, 191350. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Ihl, R.; Korczyn, A.D.; Lanza, G.; Jansa, J.; Hoerr, R.; Guekht, A. Towards the concept of disease-modifier in post-stroke or vascular cognitive impairment: A consensus report. BMC Med. 2017, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Cantone, M.; Musso, S.; Borgione, E.; Scuderi, C.; Ferri, R. Early-onset subcortical ischemic vascular dementia in an adult with mtDNA mutation 3316G>A. J. Neurol. 2018, 265, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Hannequin, D.; Coutant, S.; Rovelet-Lecrux, A.; Wallon, D.; Rousseau, S.; Legallic, S.; Paquet, C.; Bombois, S.; Pariente, J.; et al. PHRC GMAJ Collaborators. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 2012, 17, 875–879. [Google Scholar] [CrossRef]
- Caglayan, S.; Takagi-Niidome, S.; Liao, F.; Carlo, A.S.; Schmidt, V.; Burgert, T.; Kitago, Y.; Füchtbauer, E.M.; Füchtbauer, A.; Holtzman, D.M.; et al. Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci. Transl. Med. 2014, 6, 223. [Google Scholar] [CrossRef]
- Cuccaro, M.L.; Camey, R.M.; Zhang, Y.; Bohm, C.; Kunkle, B.W.; Vardarajan, B.N.; Whitehead, P.L.; Cukier, H.N.; Mayeux, R.; George-Hyslop, P.; et al. SORL1 mutations in early- and late-onset Alzheimer disease. Neurol. Genet. 2016, 2, e116. [Google Scholar]
- Nicolas, G.; Charbonnier, C.; Wallon, D.; Quenez, O.; Bellenguez, C.; Grenier-Boley, B.; Rousseau, S.; Richard, A.C.; Rovelet-Lecrux, A.; Le Guennec, K.; et al. CNR-MAJ collaborators. SORL1 rare variants: A major risk factor for familial early-onset Alzheimer’s disease. Mol. Psychiatry 2016, 21, 831–836. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Zhang, Y.; Lee, J.H.; Cheng, R.; Bohm, C.; Ghani, M.; Cheng, R.; Bohm, C.; Ghani, M.; Reitz, C.; et al. Coding mutations in SORL1 and Alzheimer disease. Ann. Neurol. 2015, 77, 215–227. [Google Scholar] [CrossRef]
- Li, X.; Xiong, Z.; Liu, Y.; Yuan, Y.; Deng, J.; Xiang, W.; Li, Z. Case report of first—Episode psychotic symptoms in a patient with early-onset Alzheimer’s disease. BMC Psychiatry 2020, 20, 128. [Google Scholar] [CrossRef]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Shen, L.; Jia, J. An overview of Genome-Wide Association Studies in Alzheimer’s Disease. Neurosci. Bull. 2016, 32, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Gen. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; Lambert, J.C.; Rogaeva, E.; Vandenberghe, R.; Le Bastard, N.; Pasquier, F.; Vermeulen, S.; Van Dongen, J.; et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol. Neurodegener. 2012, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Vermeulen, S.; Van Cauwenberghe, C.; Heeman, B.; Asselbergh, B.; Robberecht, C.; Engelborghs, S.; Vandenbulcke, M.; Vandenberghe, R.; De Deyn, P.P.; et al. Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol. Neurodegener. 2015, 10, 30. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef]
- Lue, L.F.; Schmitz, C.; Walker, D.G. What happens to microglial TREM2 in Alzheimer’s disease: Immunoregulatory turned into immunopathogenic? Neuroscience 2015, 302, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Yamanishi, Y.; Suarez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; Bettens, K.; Philtjens, S.; Van Langenhove, T.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Vandenbulcke, M.; Van Dongen, J.; Geerts, N.; et al. BELNEU consortium. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 2014, 35, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Richard, A.C.; Rollin-Sillaire, A.; Frebourg, T.; Campion, D.; Hannequin, D. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Karch, C.M.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.; Bertelsen, S.; et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in 74 ABCA7 in a Belgian cohort of Alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822. [Google Scholar] [CrossRef]
- Steinberg, S.; Stefansson, H.; Jonsson, T.; Johannsdottir, H.; Ingason, A.; Helgason, H.; Sulem, P.; Magnusson, O.T.; Gudjonsson, S.A.; Unnsteinsdottir, U.; et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat. Genet. 2015, 47, 445–447. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide 76 association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; St George-Hyslop, P.; Fraser, P.E. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef]
- Bonvicini, C.; Scassellati, C.; Benussi, L.; Di Maria, E.; Maj, C.; Ciani, M.; Fostinelli, S.; Mega, A.; Bocchetta, M.; Lanzi, G.; et al. Next Generation Sequencing Analysis in Early Onset Dementia Patients. J. Alzheimers Dis. 2019, 67, 243–256. [Google Scholar] [CrossRef]
- Cochran, J.N.; McKinley, E.C.; Cochran, M.; Amaral, M.D.; Moyers, B.A.; Lasseigne, B.N.; Gray, D.E.; Lawlor, J.M.J.; Prokop, J.W.; Geier, E.G.; et al. Genome sequencing for early-onset or atypical dementia: High diagnostic yield and frequent observation of multiple contributory alleles. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003491. [Google Scholar] [CrossRef] [PubMed]
- Cali, F.; Cantone, M.; Cosentino, F., II; Lanza, G.; Ruggeri, G.; Chiavetta, V.; Salluzzo, R.; Ragalmuto, A.; Vinci, M.; Ferri, R. Interpreting Genetic Variants: Hints from a Family Cluster of Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef]
- D’Argenio, V.; Sarnataro, D. Microbiome influence in the pathogenesis of prion and Alzheimer’s Diseases. Int. J. Mol. Sci. 2019, 20, 4704. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Song, M.Y.; Kim, D.; Park, C.; Park, D.K.; Kim, D.G.; Yoo, J.S.; Kim, Y.H. A Proteotranscriptomic-based computational drug-repositioning method for Alzheimer’s Disease. Front. Pharmacol. 2020, 10, 1653. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation and Protein Region | Protein Stability/Folding/Processing | Reference |
|---|---|---|
| N-terminal | ||
| p.K670N/p.M671L | Leads to increased absolute levels of Aβ42, doesn’t alter Aβ42/Aβ40 ratio Present in MAMs | Kumar-Singh_2009 [22] Del Prete_2017 [23] |
| Amyloid-beta domain | ||
| p.A692G | Murakami_2002 [21] | |
| p.E693Q p.E693K p.E693G | Potent aggregative | |
| p.D694N | ||
p.A713T p.T714I | Increase Aβ42/Aβ40 ratio, affect stability of APP CTFs | Kumar-Singh_2009 [22] |
| Transmembrane/C-terminal | ||
| p.V715M p.I716V p.V717L | Increase Aβ42/Aβ40 ratio, affect stability of APP CTFs | De Jonghe_2001 [20] |
| Pp.I716T | Increases Aβ42/Aβ40 ratio reducing the efficiency of the γ-secretase ε-cleavage | Götz_2019 [25] |
| p.L723P | Causes unfolding of C-terminal turn of APP TM domain helix | Bocharov_2019 [26] |
| Gene Name (Acronym) | Proposed Function in FAD | References |
|---|---|---|
| Apolipoprotein E (APOE) | Contribution of ApoE4 to mitochondrial dysfunction, cytoskeletal disorganization and neurofibrillary tangles | Huang_2006 [53] |
| Neurogenic Locus Notch Homolog Protein 3 (NOTCH3) | Cellular signaling impairment | Patel_2019 [61] |
| Sortilin-related receptor (SORL1) | Interference with APP trafficking and alteration of the Aβ levels | Cuccaro_2016 [66] |
| Complement component 3b/4b receptor 1 (CR1) | Aβ peptide clearance reduction | Shen 2016 [72] |
| Clusterin (CLU) | Synapsis turnover reduction | Bettens_2015 [75] |
| Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) | Aβ peptide clearance reduction | Bailey_2015 [77] Lue_2015 [78] Kleinberger_2014 [79] |
| The ATP-binding cassette, subfamily a, member 7 (ABCA7) | Aβ peptide production increase | Satoh_2015 [86] |
| Ephrin receptor (EPHA1) | Alteration of neuronal development | Vardarajan_2016 [85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Argenio, V.; Sarnataro, D. New Insights into the Molecular Bases of Familial Alzheimer’s Disease. J. Pers. Med. 2020, 10, 26. https://doi.org/10.3390/jpm10020026
D’Argenio V, Sarnataro D. New Insights into the Molecular Bases of Familial Alzheimer’s Disease. Journal of Personalized Medicine. 2020; 10(2):26. https://doi.org/10.3390/jpm10020026
Chicago/Turabian StyleD’Argenio, Valeria, and Daniela Sarnataro. 2020. "New Insights into the Molecular Bases of Familial Alzheimer’s Disease" Journal of Personalized Medicine 10, no. 2: 26. https://doi.org/10.3390/jpm10020026
APA StyleD’Argenio, V., & Sarnataro, D. (2020). New Insights into the Molecular Bases of Familial Alzheimer’s Disease. Journal of Personalized Medicine, 10(2), 26. https://doi.org/10.3390/jpm10020026