Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)

 ,
, 
Abstract
1. Introduction
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Significant Global Health Problem
2. Clinical Characteristics of ME/CFS
3. Clinical Case Definitions
4. Physiological Cause of ME/CFS and Current Treatments
5. Biomarkers Leading to a Diagnostic Test
6. Changes in the Biology of ME/CFS Patients
7. Global Research into ME/CFS Biology
7.1. Recent Research Studies Have Focused on Several Key Areas
7.1.1. Microbiome
7.1.2. Metabolome
7.1.3. Mitochondria
7.1.4. Transient Receptor Potential (TRP) Ion Channels
7.1.5. Genetic Susceptibility
8. Has Biomedical Research Informed the Clinic, and Assisted Diagnosis and Treatment?
9. Discussion
Future Directions and Unresolved Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bansal, A.S.; Bradley, A.S.; Bishop, K.N.; Kiani-Alikhan, S.; Ford, B. Chronic fatigue syndrome, the immune system and viral infection. Brain Behav. Immun. 2012, 26, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hunt, G. $3 Million Research Funding for Chronic Fatigue Syndrome; Media Release; Australian Government Department of Health: Canberra, Australia, 27 March 2019. Available online: https://beta.health.gov.au/ministers/the-hon-greg-hunt-mp/media/3-million-research-funding-for-chronic-fatigue-syndrome (accessed on 10 July 2019).
- Associated New Zealand ME Society (ANZMES). Available online: http://anzmes.org.nz/ (accessed on 4 April 2019).
- Simpson, L.O. Myalgic Encephalomyelitis. J. R. Soc. Med. 1991, 84, 633. [Google Scholar] [PubMed]
- Price, J.L. Myalgic encephalomyelitis. Lancet 1961, 1, 737–738. [Google Scholar] [CrossRef]
- IOM (Institute of Medicine). Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Johnston, S.; Brenu, E.W.; Staines, D.; Marshall-Gradisnik, S. The prevalence of chronic fatigue syndrome/myalgic encephalomyelitis: A meta analysis. Clin. Epidemiol. 2013, 5, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.D.; Antoni, M.H.; Fletcher, M.A.; Maher, K.; Segota, M.C.; Klimas, N. Impaired natural immunity, cognitive dysfunction, and physical symptoms in patients with chronic fatigue syndrome: Preliminary evidence for a subgroup? J. Psychosom. Res. 2006, 60, 559–566. [Google Scholar] [CrossRef]
- Lorusso, L.; Mikhaylova, S.V.; Capelli, E.; Ferrari, D.; Ngonga, G.K.; Ricevuti, G. Immunological aspects of chronic fatigue syndrome. Autoimmun. Rev. 2009, 8, 287–291. [Google Scholar] [CrossRef]
- Devanur, L.D.; Kerr, J.R. Chronic fatigue syndrome. J. Clin. Virol. 2006, 37, 139–150. [Google Scholar] [CrossRef]
- Holgate, S.T.; Komaroff, A.L.; Mangan, D.; Wessely, S. Chronic fatigue syndrome: Understanding a complex illness. Nat. Rev. Neurosci. 2011, 12, 539–544. [Google Scholar] [CrossRef]
- Crawley, E. The epidemiology of chronic fatigue syndrome/myalgic encephalitis in children. Arch. Dis. Child. 2014, 99, 171–174. [Google Scholar] [CrossRef]
- Vallings, R. Chronic Fatigue Syndrome M.E. Symptoms: Diagnosis & Management; Calico Publishing Ltd.: Auckland, New Zealand, 2012. [Google Scholar]
- Buchwald, D.; Herrell, R.; Ashton, S.; Belcourt, M.; Schmaling, K.; Sullivan, P.; Neale, M.; Goldberg, J. A twin study of chronic fatigue. Psychosom. Med. 2001, 63, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Sabath, D.E.; Barcy, S.; Koelle, D.M.; Zeh, J.; Ashton, S.; Buchwald, D. Cellular immunity in monozygotic twins discordant for chronic fatigue syndrome. J. Infect. Dis. 2002, 185, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Evengard, B.; Jacks, A.; Pedersen, N.L. Twin analyses of chronic fatigue in a Swedish national sample. Psychol. Med. 2005, 35, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Chaudhuri, A. ME/CFS/PVFS: An Exploration of the Key Clinical Issues, 9th ed.; The ME Association: Gawcott, UK, 2017. [Google Scholar]
- Agmon-Levin, N.; Zafrir, Y.; Kivity, S.; Balofsky, A.; Amital, H.; Shoenfeld, Y. Chronic fatigue syndrome and fibromyaligia following immunization with the hepatitis B vaccine: Another angle of the ‘autoimmune (auto-inflammatory) syndrome induced by adjuvants’ ASIA. Immun. Res. 2014, 60, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, F.C.; Bateman, L.; Bested, A.C.; Davenport, T.; Friedman, K.; Gurwitt, A.; Jason, L.A.; Lapp, C.W.; Stevens, S.R.; Underhill, R.A.; et al. Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: A Primer for Clinical Practitioners; International Association for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: Chicago, IL, USA, 2012. [Google Scholar]
- Cairns, R.; Hotopf, M. A systematic review describing the prognosis of chronic fatigue syndrome. Occup. Med. 2005, 55, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Brurberg, K.G.; Fonhus, M.S.; Larun, L.; Flottorp, S. Case definitions for chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME): A systematic review. BMJ Open 2014, 4, 1–12. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Kumar Jain, A.; de Meirleir, K.L.; Peterson, L.; Klimas, N.G.; Lerner, M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Guidelines, A Consensus Document. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Johnston, S.; Brenu, E.; Staines, D.; Marshall-Gradisnik, S. The role of clinical guidelines for chronic fatigue syndrome/myalgic encephalomyelitis in research settings. Fatigue 2014, 2, 28–39. [Google Scholar] [CrossRef]
- Whiting, P.; Bagnall, A.M.; Sowden, A.J.; Cornell, J.E.; Mulrow, C.D.; Ramírez, G. Interventions for the treatment and management of chronic fatigue syndrome: A systematic review. JAMA 2001, 286, 1360–1368. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O. Clinical impact of B-cell depletion with the anti-CD20 antibody rituximab in chronic fatigue syndrome: A preliminary case series. BMC Neurol. 2009, 9, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Fluge, O.; Bruland, O.; Risa, K.; Storstein, A.; Kristoffersen, E.K.; Sapkota, D.; Naess, H.; Dahl, O.; Nyland, H.; Mella, O. Benefit from B-lymphocyte depletion using the anti-CD20 antibody rituximab in chronic fatigue syndrome. A double-blind and placebo-controlled study. PLoS ONE 2011, 6, e26358. [Google Scholar] [CrossRef] [PubMed]
- Fluge, O.; Risa, K.; Lunde, S.; Alme, K.; Gurvin Rekeland, I.; Sapkota, D.; Kleboe Kristofferson, E.; Sorland, K.; Bruland, O.; Dahl, O.; et al. B-Lymphocyte depletion in myalgic encephalopathy/chronic fatigue syndrome. an open-label phase II study with rituximab maintenance treatment. PLoS ONE 2015, 10, 0129898. [Google Scholar] [CrossRef] [PubMed]
- Maxmen, A. A reboot for chronic fatigue syndrome research. Nature 2018, 553, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Anderson, J.N.; Adolphs, D.L.; Bateman, L.; Klimas, N.; Levine, S.M.; Garvert, D.; Kaiser, J.D. KPAX002 as a treatment for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): A prospective, randomized trial. Int. J. Clin. Exp. Med. 2018, 11, 2890–2900. [Google Scholar]
- Sharpe, M.; Goldsmith, K.A.; Johnson, A.L.; Chalder, T.; Walker, J.; White, P.D. Rehabilitative treatments for chronic fatigue syndrome: Long-term follow-up from the PACE trial. Lancet Psychiatry 2015, 2, 1067–1074. [Google Scholar] [CrossRef]
- Coyne, J.C.; Laws, K.R. Results of the PACE follow-up study are uninterpretable. Lancet Psychiatry 2016, 3, e6–e7. [Google Scholar] [CrossRef]
- Marks, D.F. Special issue: The PACE Trial. J. Health Psychol. 2017, 22, 1103–1216. [Google Scholar] [CrossRef]
- Esfandyarpour, R.; Kashi, A.; Nemat-Gorgani, M.; Wilhelmy, J.; Davis, R.W. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc. Natl. Acad. Sci. USA 2019, 116, 10250–10257. [Google Scholar] [CrossRef]
- Tate, W.P.; Sweetman, E.C.; Noble, A.J.K.; Edgar, C.; Bateman, G.; Mackay, A.; Ryan, M.; Hodges, L.; Vallings, R. Tackling ME/CFS in New Zealand by the principles of precision medicine. IiME 2016, 10, 46–55. [Google Scholar]
- Noble, A.J.K. Exploring Potential Biomarkers for ME/CFS. Ph.D. Thesis, University of Otago, Dunedin, New Zealand, 2017. [Google Scholar]
- Sweetman, E.C. Comprehensive Molecular Analysis of Different Classes of Molecules in a Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Pilot Study Group, and Investigation of RNA-activated Protein Kinase R (PKR) as a Diagnostic Biomarker. Ph.D. Thesis, University of Otago, Dunedin, New Zealand, 2018. [Google Scholar]
- Gow, J.W.; Hagan, S.; Herzyk, P.; Cannon, C.; Behan, P.O.; Chaudhuri, A. A gene signature for post-infectious chronic fatigue syndrome. BMC Med. Genom. 2009, 2, 38–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brenu, E.W.; Ashton, K.J.; Batovska, J.; Staines, D.R.; Marshall-Gradisnik, S.M. High-throughput sequencing of plasma microRNA in chronic fatigue syndrome/myalgic encephalomyelitis. PLoS ONE 2014, 9, e102783. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.B.; Alsøe, L.; Lindvall, J.M.; Sulheim, D.; Fagermoen, E.; Winger, A.; Kaarbo, M.; Nilsen, H.; Wyller, V.B. Whole blood gene expression in adolescent chronic fatigue syndrome: An exploratory cross-sectional study suggesting altered B cell differentiation and survival. J. Transl. Med. 2017, 15, 102–122. [Google Scholar] [CrossRef] [PubMed]
- Broderick, G.; Katz, B.Z.; Fernandes, H.; Fletcher, M.A.; Klimas, N.; Smith, F.A.; O’Gorman, M.R.; Vernon, S.D.; Taylor, R. Cytokine expression profiles of immune imbalance in post-mononucleosis chronic fatigue. J. Transl. Med. 2012, 10, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Drystad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. The association of fecal microbiota and fecal, blood serum and urine metabolites in myalgic encephalomyelitis/chronic fatigue syndrome. Metabolomics 2017, 13, 8–20. [Google Scholar] [CrossRef]
- de Vega, W.C.; Herrera, S.; Vernon, S.D.; McGowan, P.O. Epigenetic modifications and glucocorticoid sensitivity in myalgic encephalomyelitis/chronic fatigue syndrome. BMC Med. Genom. 2017, 10, 11–24. [Google Scholar] [CrossRef]
- Sweetman, E.C.; Ryan, M.; Edgar, C.; Mackay, A.; Vallings, R.; Tate, W. Changes in the transcriptome of circulating immune cells of a New Zealand cohort with Myalgic encephalomyelitis/chronic fatigue syndrome. Int. J. Immunopathol. Pharmacol. 2019, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.K.; Jayanthan, R.K.; Mitchell, G.W.; Carreras Tartak, J.A.; Croglio, M.P.; Suarez, A.; Liu, A.Y.; Razzo, B.M.; Oyeniran, E.; Ruth, J.R.; et al. Taking control of castleman disease: Leveraging precision medicine technologies to accelerate rare disease research. Yale J. Biol. Med. 2015, 88, 383–388. [Google Scholar] [PubMed]
- Lee, R.E.C.; Walker, S.R.; Savery, K.; Frank, D.A.; Gaudet, S. Fold-change of nuclear NF-κB determines TNF-induced transcription in single cells. Mol. Cell 2014, 53, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Meeus, M.; Nijs, J.; McGregor, N.; Meeusen, R.; De Schutter, G.; Truijen, S.; Fremont, M.; Van Hoof, E.; De Meirler, K. Unravelling intracellular immune dysfunctions in chronic fatigue syndrome: Interactions between protein kinase R activity, RNase L cleavage and elastase activity, and their clinical relevance. In Vivo 2008, 22, 115–122. [Google Scholar] [PubMed]
- Shukla, S.K.; Cook, D.; Meyer, J.; Vernon, S.D.; Le, T.; Clevidence, D.; Robertson, C.E.; Schrodi, S.J.; Yale, S.; Frank, D.N. Changes in gut and plasma microbiome following exercise challenge in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). PLoS ONE 2015, 10, e0145453. [Google Scholar] [CrossRef]
- Mandarano, A.H.; Giloteaux, L.; Keller, B.A.; Levine, S.M.; Hanson, M.R. Eukaryotes in the gut microbiota in myalgic encephalomyelitis/chronic fatigue syndrome. PeerJ 2018, 6, e4282. [Google Scholar] [CrossRef]
- Maes, M.; Mihaylova, I.; Leunis, J.C. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): Indication for the involvement of gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J. Affect. Disord. 2007, 99, 237–240. [Google Scholar] [CrossRef]
- Giloteaux, L.; Hanson, M.R.; Keller, B.A. A pair of identical twins discordant for myalgic encephalomyelitis/chronic fatigue syndrome differ in physiological parameters and gut microbiome composition. Am. J. Case Rep. 2016, 17, 720–729. [Google Scholar] [CrossRef]
- Giloteaux, L.; Goodrich, J.K.; Walters, W.A.; Leine, S.M.; Ley, R.E.; Hanson, M.R. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2016, 4, 30–41. [Google Scholar] [CrossRef]
- Ellis, J.E.; Missan, D.S.; Shabilla, M.; Martinez, D.; Fry, S.E. Microbial community profiling of peripheral blood in myalgic encephalomyelitis/chronic fatigue syndrome. Hum. Microbiome J. 2018, 9, 16–21. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Williams, B.L.; Mishra, N.; Che, X.; Lee, B.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; Levine, S.; Montoya, J.G.; et al. Fecal metagenomics profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 2017, 5, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. Metabolic profiling reveals anomalous energy metabolism and oxidative stress pathways in chronic fatigue syndrome patients. Metabolomics 2015, 11, 1626–1639. [Google Scholar] [CrossRef]
- Yamano, E.; Sugimoto, M.; Hirayama, A.; Kume, S.; Yamato, M.; Jin, G.; Tajima, S.; Goda, N.; Iwai, K.; Fukuda, S.; et al. Index markers of chronic fatigue syndrome with dysfunction of TCA and urea cycles. Sci. Rep. 2016, 6, 34990–34998. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.H.; Derrington, I.M.; Brinkerhoff, H.; Lanford, K.W.; Nova, I.C.; Samson, J.M.; Bartlett, J.j.; Pavlenok, M.; Gundlach, J.H. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. USA 2013, 110, 18904–18909. [Google Scholar] [CrossRef] [PubMed]
- de Vega, W.C.; Erdman, L.; Vernon, S.D.; Goldenberg, A.; McGowan, P.O. Integration of DNA methylation and health scores identifies subtypes in myalgic encephalomyelitis/chronic fatigue syndrome. Epigenomics 2018, 10, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Herrera, S.; de Vega, W.C.; Ashbrook, D.; Vernon, S.D.; McGowan, P.O. Genome-epigenome interactions associated with myalgic encephalomyelitis/chronic fatigue syndrome. Epigenetics 2018, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- de Vega, W.C.; Vernon, S.D.; McGowan, P.O. DNA methylation modifications associated with chronic fatigue syndrome. PLoS ONE 2014, 9, e104757. [Google Scholar] [CrossRef] [PubMed]
- Brenu, E.W.; Staines, D.R.; Marshall-Gradisnik, S. Methylation profile of CD4+ T cells in chronic fatigue syndrome/myalgic encephalomyelitis. J. Clin. Cell Immunol. 2014, 5, 228–241. [Google Scholar]
- Gorman, G.S.; Elson, J.L.; Newman, J.; Payne, B.; McFarland, R.; Newton, J.L.; Turnbull, D.M. Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul. Disord. 2015, 25, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Staines, D.R.; Du Preez, S.; Cabanas, H.; Balinas, C.; Eaton, N.; Passmore, R.; Maksoud, R.; Redmayne, J.; Marshall-Gradisnik, S. Transient receptor potential ion channels in the etiology and pathomechanism of chronic fatigue syndrome/myalgic encephalomyelitis. Int. J. Clin. Med. 2018, 9, 445–453. [Google Scholar] [CrossRef][Green Version]
- Nguyen, T.; Johnston, S.C.; Clarke, L.; Smith, P.; Marshall-Gradisnik, S. Imparied calcium mobilization in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients is associated with transient receptor potential melastatin 3 ion channels. Clin. Exp. Immunol. 2017, 187, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Staines, D.R.; Nilius, B.; Smith, P.; Marshall-Gradisnik, S. Novel identification and characterisation of transient receptor potential melastatin 3 ion channels on natural killer cells and b lymphocytes: Effects on cell signalling in chronic fatigue syndrome/myalgic encephalomyelitis. Biol. Res. 2016, 49, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Marshall-Gradisnik, S.; Johnston, S.; Chacko, A.; Nguyen, T.; Smith, P.; Staines, D. Single nucleotide polymorphisms and genotypes of transient receptor potential ion channel and acetylcholine receptor genes from isolated b lymphocytes in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Int. Med. Res. 2016, 44, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, H.; Muraki, K.; Eaton, N.; Balinas, C.; Staines, D.; Marshall-Gradisnik, S. Loss of transient receptor potential melastatin 3 ion channel function in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients. Mol. Med. 2018, 24, 44–53. [Google Scholar] [CrossRef]
- Held, K.; Voets, T.; Vriens, J. TRPM3 in temperature sensing and beyond. Temperature 2015, 2, 201–213. [Google Scholar] [CrossRef]
- Smith, A.K.; Fang, H.; Whistler, T.; Unger, E.R.; Rajeevan, M.S. Convergent genomic studies identify association of GRIK2 and NPAS2 with chronic fatigue syndrome. Neuropsychobiology 2011, 64, 183–194. [Google Scholar] [CrossRef]
- Schlauch, K.A.; Khaiboullina, S.F.; De Meirleir, K.L.; Rawat, S.; Petereit, J.; Rizvanov, A.A.; Blatt, N.; Mijatovic, T.; Kulick, D.; Palotas, A.; et al. Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome. Transl. Psychiatry 2016, 6, e730. [Google Scholar] [CrossRef]
- World Health Organization. International Classification of Diseases, Eighth Revision (ICD-8): I (Code 323): 158; World Health Organization: Geneva, Switzerland, 1967. [Google Scholar]
- Mackay, A.; Tate, W.P. A compromised paraventricular nucleus within a dysfunctional hypothalamus: A novel neuroinflammatory paradigm for ME/CFS. Int. J. Immunopathol. Pharmacol. 2018, 32, 1–8. [Google Scholar] [CrossRef]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; Wada, Y.; Tanaka, M.; Tazawa, S.; Onoe, K.; Fukuda, S.; Kawabe, J.; Takahashi, K.; et al. Neuroinflammation in patients with chronic fatigue syndrome/Myalgic encephalomyelitis: An 11C-(R)-PK11195 PET study. J. Nucl. Med. 2014, 55, 945–950. [Google Scholar] [CrossRef]



{kind=link}
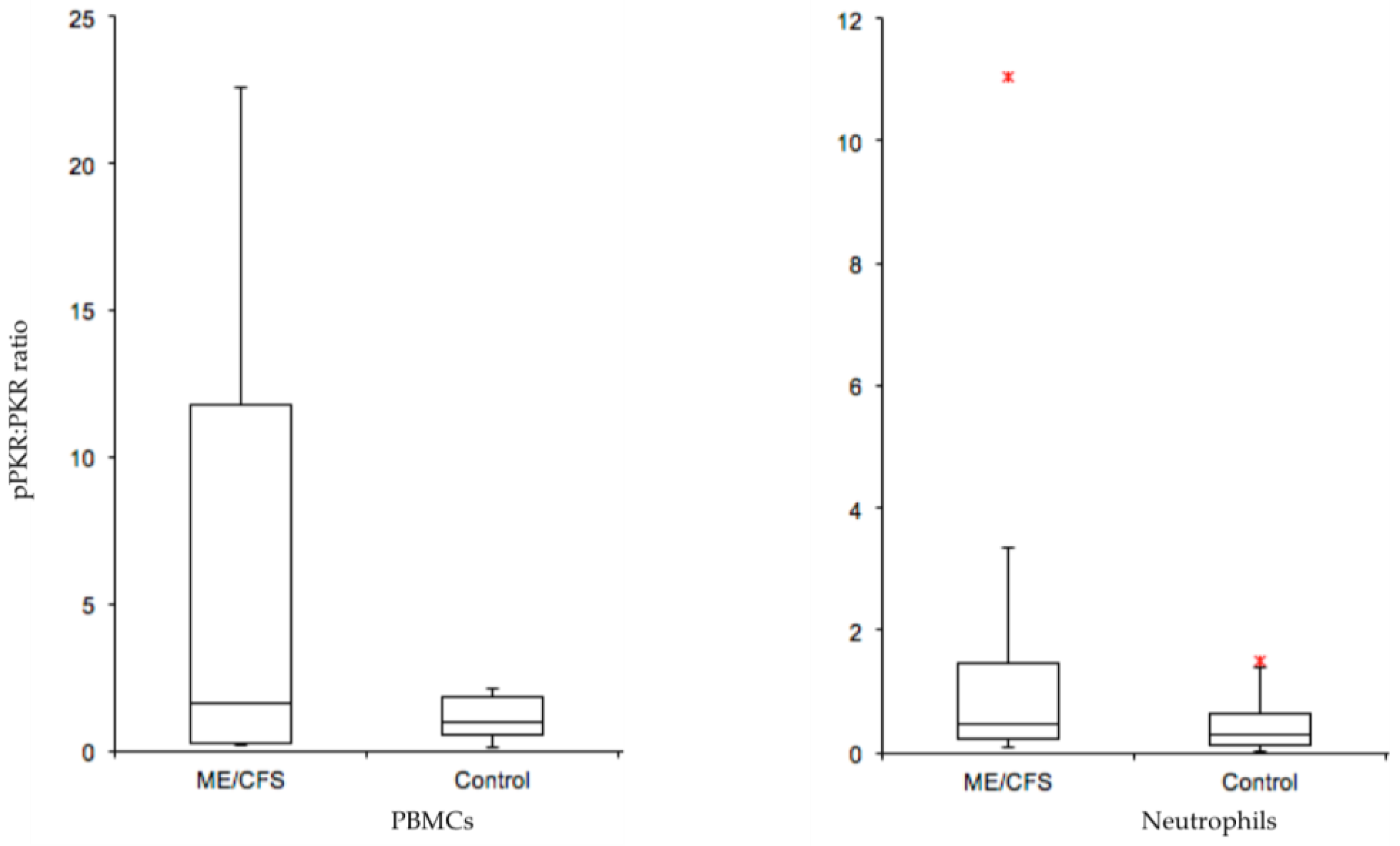{kind=link}
| Affected Biological Pathways | References |
|---|---|
| Immune/inflammation | [37,38,39,40,41,49] |
| Cytokine regulation | [37,38,42,43,49] |
| Metabolic dysregulation | [38,44,45,49] |
| Mitochondrial dysfunction | [38,45,46,49] |
| Oxidative stress | [38,39,47,49] |
| Apoptosis | [38,39,47,49] |
| Circadian rhythm | [48,49] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweetman, E.; Noble, A.; Edgar, C.; Mackay, A.; Helliwell, A.; Vallings, R.; Ryan, M.; Tate, W. Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Diagnostics 2019, 9, 73. https://doi.org/10.3390/diagnostics9030073
Sweetman E, Noble A, Edgar C, Mackay A, Helliwell A, Vallings R, Ryan M, Tate W. Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Diagnostics. 2019; 9(3):73. https://doi.org/10.3390/diagnostics9030073
Chicago/Turabian StyleSweetman, Eiren, Alex Noble, Christina Edgar, Angus Mackay, Amber Helliwell, Rosamund Vallings, Margaret Ryan, and Warren Tate. 2019. "Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)" Diagnostics 9, no. 3: 73. https://doi.org/10.3390/diagnostics9030073
APA StyleSweetman, E., Noble, A., Edgar, C., Mackay, A., Helliwell, A., Vallings, R., Ryan, M., & Tate, W. (2019). Current Research Provides Insight into the Biological Basis and Diagnostic Potential for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Diagnostics, 9(3), 73. https://doi.org/10.3390/diagnostics9030073