Hybrid Benign Peripheral Nerve Sheath Tumors: A Comprehensive Literature Review with Emphasis on Their Clinical, Morphological and Genetic Features

 ,
,  ,
,  ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion and Exclusion Criteria
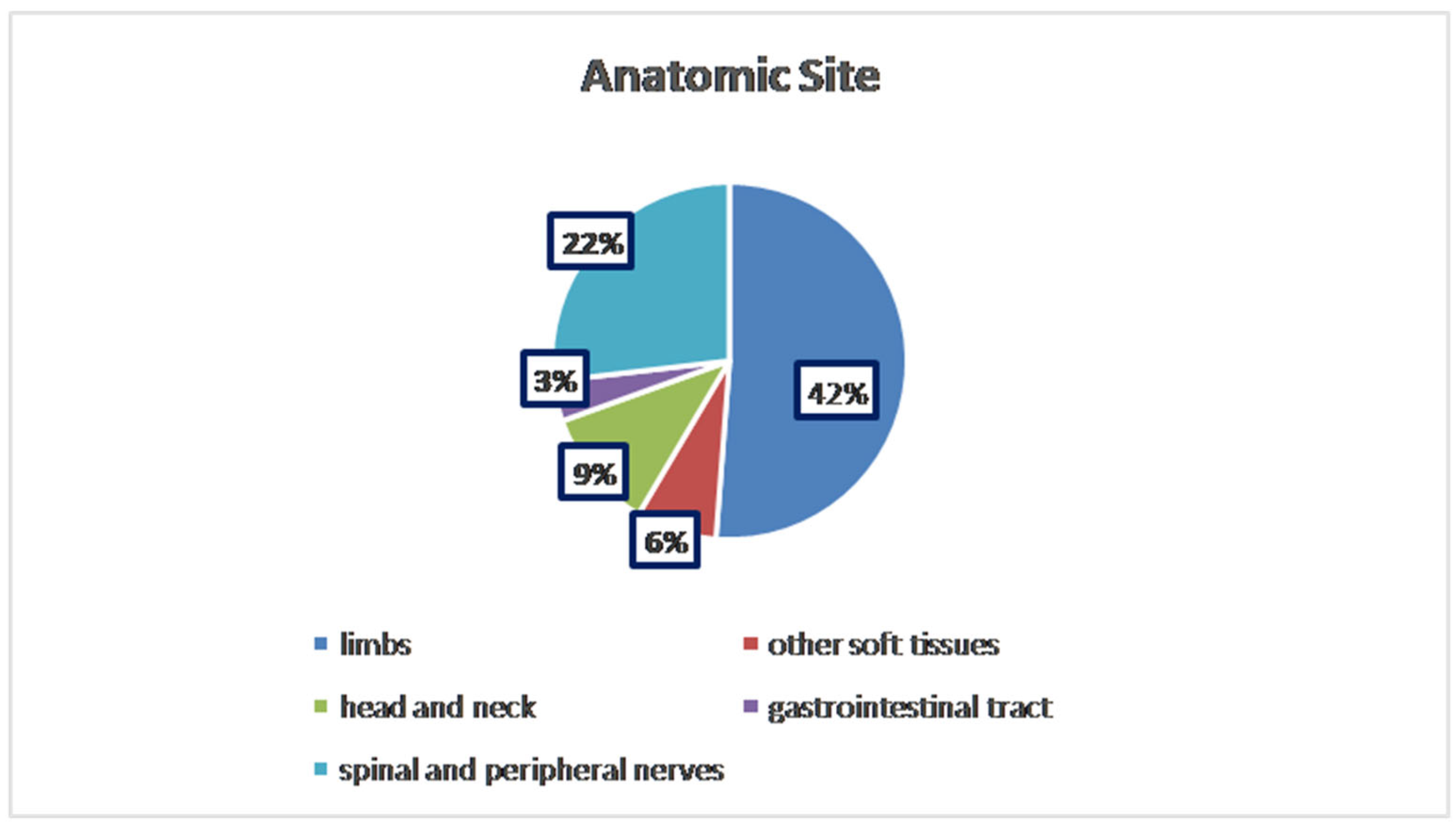
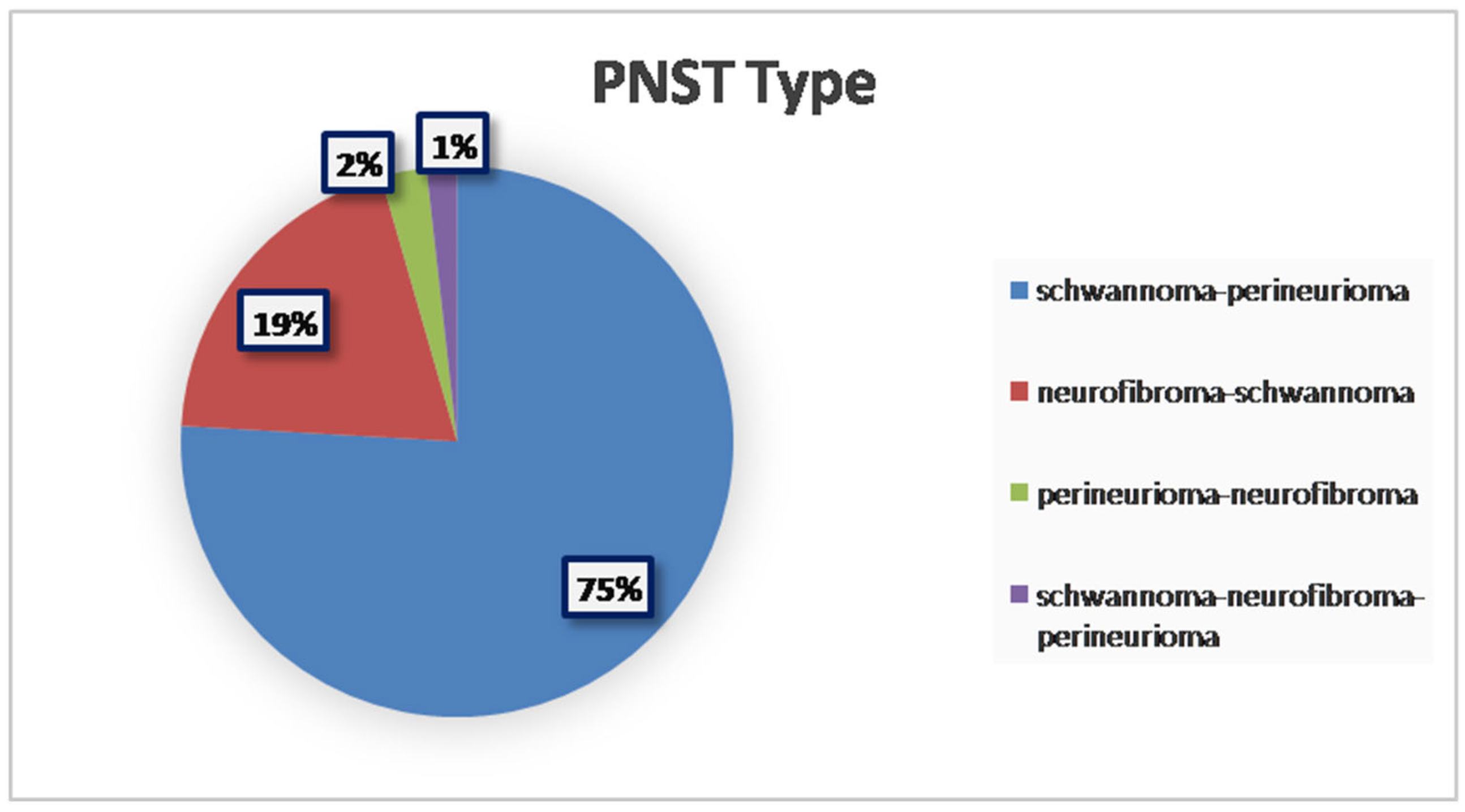
3. Results
4. Discussion
4.1. Schwannoma–Perineurioma
4.2. Schwannoma–Neurofibroma
4.3. Perineurioma–Neurofibroma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HPNSTs | Hybrid Peripheral Nerve Sheath Tumors |
| WHO | World Health Organization |
| PNSTs | Peripheral nerve sheath tumors |
| MPNSTs | Malignant peripheral nerve sheath tumors |
| MeSH | Medical Subject Headings |
| IHC | Immunohistochemical |
| NF1 | Neurofibromatosis type 1 |
| BCPHTPCN | Benign cutaneous plexiform hybrid tumor of perineurioma and cellular neurothekeoma |
References
- Central Nervous System Tumors, 5th ed.; WHO Press: Geneva, Switzerland, 2021.
- Soft Tissue and Bone Tumors, 5th ed.; WHO Press: Geneva, Switzerland, 2020.
- Michal, M.; Kazakov, D.V.; Belousova, I.; Bisceglia, M.; Zamecnik, M.; Mukensnabl, P. A benign neoplasm with histopathological features of both schwannoma and retiform perineurioma (benign schwannoma-perineurioma): A report of six cases of a distinctive soft tissue tumor with a predilection for the fingers. Virchows Arch. 2004, 445, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.C. Primary intraosseous hybrid nerve sheath tumor of femur: A hitherto undescribed occurrence in bone with secondary aneurysmal bone cyst formation resulting in pathological fracture. Pathol. Res. Pract. 2015, 211, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Broggi, G.; Angelico, G.; Puzzo, L.; Vecchio, G.M.; Virzì, V.; Salvatorelli, L.; Ruggieri, M. Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update. Diagnostics 2022, 12, 1463. [Google Scholar] [CrossRef]
- Katsumi, T.; Hayashi, R.; Takei, S.; Ansai, O.; Takatsuka, S.; Takenouchi, T.; Saito, K.; Suda, K.; Yoshihara, K.; Nagai, T.; et al. Reevaluating hybrid neurofibroma/schwannoma: Predominance of schwannoma features despite CD34 positivity and initial neurofibroma diagnosis. J. Dermatol. 2024, 51, 1461–1469. [Google Scholar] [CrossRef]
- Feany, M.B.; Anthony, D.C.; Fletcher, C.D. Nerve sheath tumours with hybrid features of neurofibroma and schwannoma: A conceptual challenge. Histopathology 1998, 32, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kacerovska, D.; Michal, M.; Kuroda, N.; Tanaka, A.; Sima, R.; Denisjuk, N.; Kreuzberg, B.; Ricarova, R.; Kazakov, D.V. Hybrid peripheral nerve sheath tumors, including a malignant variant in type 1 neurofibromatosis. Am. J. Dermatopathol. 2013, 35, 641–649. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef]
- Agaimy, A.; Michal, M. Hybrid schwannoma-perineurioma of the gastrointestinal tract: A clinicopathologic study of 2 cases and reappraisal of perineurial cells in gastrointestinal schwannomas. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 454–459. [Google Scholar] [CrossRef]
- Bergamini, M.; Costa, A.; Noberto, L.; Torres, G.; Soares, H.; Martins, F.; de Souza, S.; Braz-Silva, P. Primary intra-osseous Hybrid Schwannoma-Perineurioma in the mandible. J. Clin. Exp. Dent. 2020, 12, e888–e891. [Google Scholar] [CrossRef]
- Lang, Y.; Liu, D.; Xiang, P.; Wang, J.; Li, Y. Primary intraosseous hybrid epithelioid schwannoma/perineurioma in the proximal tibia: A case report of benign hybrid neoplasm with local hypercellularity. Diagn. Pathol. 2019, 14, 51. [Google Scholar] [CrossRef]
- Hornick, J.L.; Bundock, E.A.; Fletcher, C.D.M. Hybrid schwannoma/perineurioma: Clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am. J. Surg. Pathol. 2009, 33, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Choong, P.; Ali, M.; Lindsay, D.; Saifuddin, A. Hybrid peripheral nerve sheath tumours: MRI features with pathological correlation in 24 cases. Br. J. Radiol. 2023, 97, 126–134. [Google Scholar] [CrossRef]
- Din, N.U.; Ahmad, Z.; Abdul-Ghafar, J.; Ahmed, R. Hybrid peripheral nerve sheath tumors: Report of five cases and detailed review of literature. BMC Cancer 2017, 17, 349. [Google Scholar] [CrossRef]
- Kuroda, N.; Kazakov, D.V.; Hes, O.; Michal, M.; Goda, M.; Miyazaki, K.; Hayashi, Y.; Okamoto, S.; Lee, G.-H. Hybrid peripheral nerve sheath tumor of the nasal cavity showing schwannomatous, neurofibromatous, and perineuriomatous areas. Med. Mol. Morphol. 2010, 43, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Chijiiwa, Y.; Sano, J.; Okamura, K.; Nishio, J. Intramuscular Hybrid Nerve Sheath Tumor of the Thigh: Case Report and Literature Review. In Vivo 2024, 38, 971–974. [Google Scholar] [CrossRef]
- Leite, A.A.; Mariz, B.A.L.A.; Oliveira, L.A.; Júnior, J.N.R.A.; de Almeida, O.P.; Vargas, P.A. Hybrid Neurofibroma/Schwannoma of the Oral Cavity: A Rare Case Report and Literature Review. Int. J. Surg. Pathol. 2023, 31, 695–701. [Google Scholar] [CrossRef]
- Emanuel, P.; Pertsemlidis, D.S.; Gordon, R.; Xu, R. Benign hybrid perineurioma-schwannoma in the colon. A case report. Ann. Diagn. Pathol. 2006, 10, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Colazo, J.M.; Perez, A.N.; Judice, A.D.; Quirion, J.; Prieto-Granada, C.N.; Holt, G.E. Benign Neurofibroma/Schwannoma Hybrid Peripheral Nerve Sheath Tumor of the Ulnar Nerve Harboring a Metastatic Papillary Thyroid Carcinoma Deposit: A Case Report of Tumor-to-Tumor Metastasis. Case Rep. Pathol. 2022, 2022, 9038222. [Google Scholar] [CrossRef]
- Goyal-Honavar, A.; Gupta, A.; Chacko, G.; Chacko, A.G. Trigeminal hybrid nerve sheath tumor—A case report and literature review. Br. J. Neurosurg. 2023, 37, 1326–1329. [Google Scholar] [CrossRef]
- Hong, S.; Hara, T. Hybrid nerve sheath tumor in the orbit: A case report and review of literature. Surg. Neurol. Int. 2019, 10, 250. [Google Scholar] [CrossRef]
- Mitsui, Y.; Ishida, E.; Ogawa, K.; Fukumoto, T.; Asada, H. Hybrid Epithelioid Schwannoma/Neurofibroma: A Report of 3 Cases. Am. J. Dermatopathol. 2025, 47, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Inatomi, Y.; Ito, T.; Nagae, K.; Yamada, Y.; Kiyomatsu, M.; Nakano-Nakamura, M.; Uchi, H.; Oda, Y.; Furue, M. Hybrid perineurioma-neurofibroma in a patient with neurofibromatosis type 1, clinically mimicking malignant peripheral nerve sheath tumor. Eur. J. Dermatol. 2014, 24, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Harder, A.; Wesemann, M.; Hagel, C.; Schittenhelm, J.; Fischer, S.; Tatagiba, M.; Nagel, C.; Jeibmann, A.; Bohring, A.; Mautner, V.-F.; et al. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am. J. Surg. Pathol. 2012, 36, 702–709. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, C.T.; Kaffenberger, B.H.; Gru, A.A. A hybrid tumor with schwannoma-perineurioma-neurofibroma morphology. J. Cutan. Pathol. 2015, 42, 911–913. [Google Scholar] [CrossRef]
- Evans, D.G.; Bowers, N.L.; Tobi, S.; Hartley, C.; Wallace, A.J.; King, A.T.; Lloyd, S.K.W.; Rutherford, S.A.; Hammerbeck-Ward, C.; Pathmanaban, O.N.; et al. Schwannomatosis: A genetic and epidemiological study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.C.; Antonescu, C.R.; Demicco, E.G.; Leong, I.; Anderson, N.D.; Swanson, D.; Zhang, L.; Fletcher, C.D.M.; Hornick, J.L. Hybrid schwannoma–perineurioma frequently harbors VGLL3 rearrangement. Mod. Pathol. 2021, 34, 1116–1124. [Google Scholar] [CrossRef]
- Nihous, H.; Baud, J.; Azmani, R.M.; Michot, A.; Perret, R.; Mayeur, L.B.; de Pinieux, G.; Milin, S.; Angot, E.; Duquenne, S.; et al. Clinicopathologic and Molecular Study of Hybrid Nerve Sheath Tumors Reveals Their Common Association With Fusions Involving VGLL3. Am. J. Surg. Pathol. 2022, 46, 591–602. [Google Scholar] [CrossRef]
- Linos, K.; Stuart, L.; Goncharuk, V.; Edgar, M. Benign cutaneous biphasic hybrid tumor of perineurioma and cellular neurothekeoma: A case report expanding the clinical and histopathologic features of a recently described entity. Am. J. Dermatopathol. 2015, 37, 319–322. [Google Scholar] [CrossRef]
- Requena, L.; Sitthinamsuwan, P.; Fried, I.; Kaddu, S.; Schirren, C.G.; Schärer, L.; Hantschke, M.; Cerroni, L.; McCalmont, T.H.; Kutzner, H. A benign cutaneous plexiform hybrid tumor of perineurioma and cellular neurothekeoma. Am. J. Surg. Pathol. 2013, 37, 845–852. [Google Scholar] [CrossRef]
- Yamada, S.; Kitada, S.; Nabeshima, A.; Noguchi, H.; Sasaguri, Y.; Hisaoka, M. Benign cutaneous plexiform hybrid tumor of perineurioma and cellular neurothekeoma arising from the nose. Diagn. Pathol. 2013, 8, 165. [Google Scholar] [CrossRef]
- Kao, E.Y.; Kernig, M.L. Neurothekeoma. In StatPearls [Internet]. (2022); StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar] [PubMed]
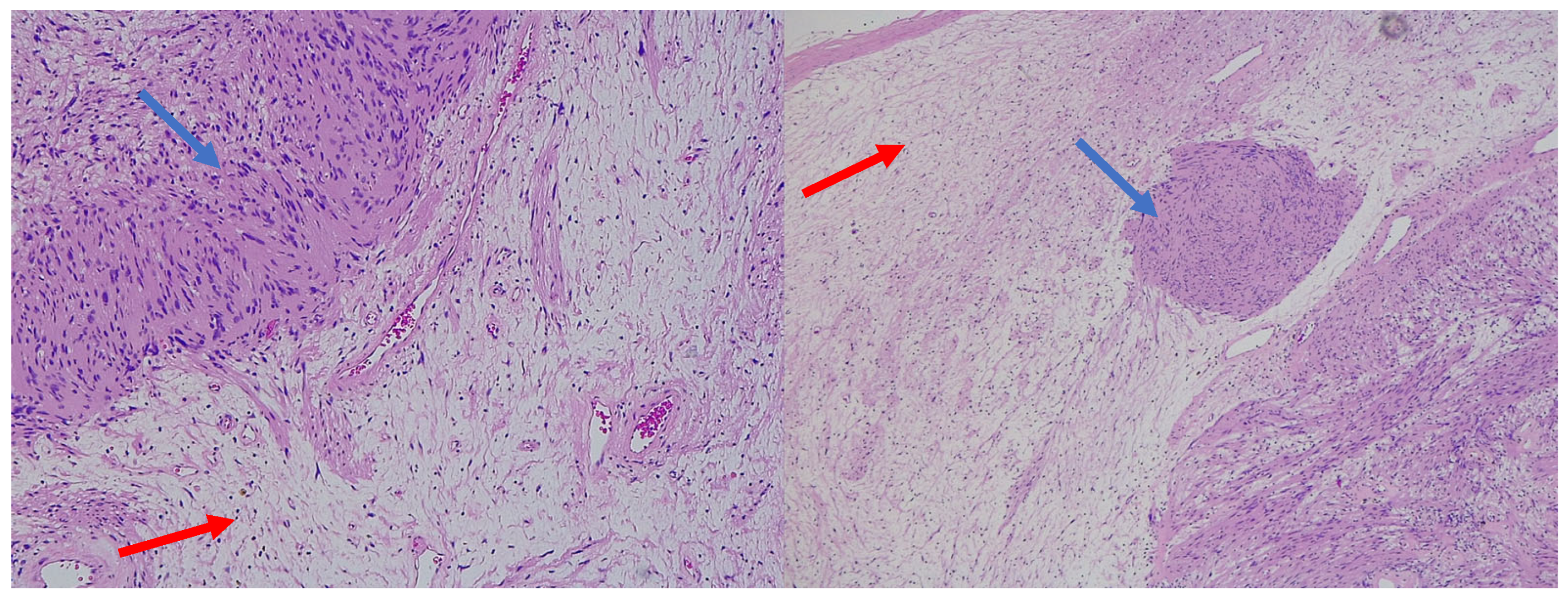
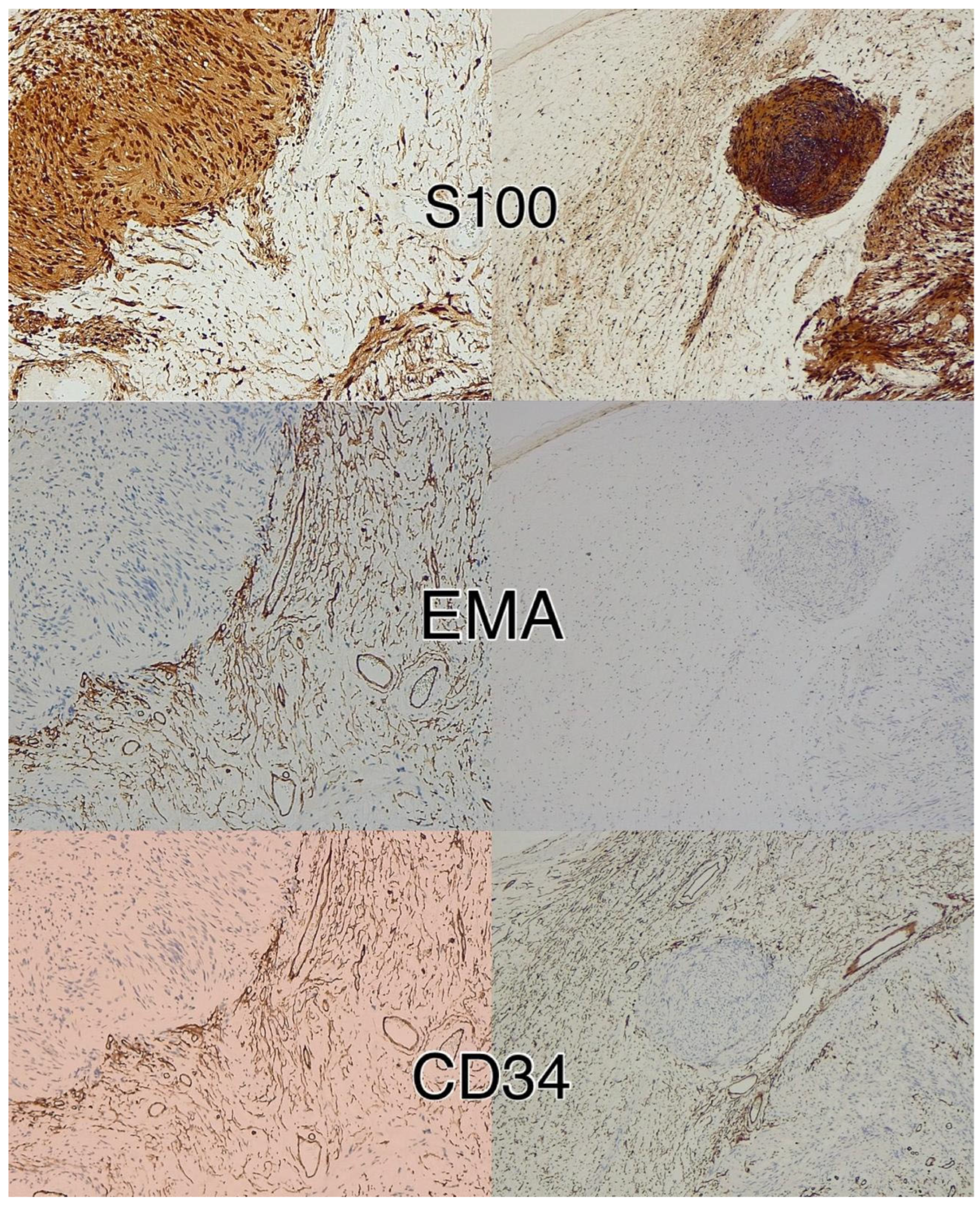

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epidemiology | Sites | Histopathology | IHC | Genetic Associations | Tumor Syndromes | |
|---|---|---|---|---|---|---|
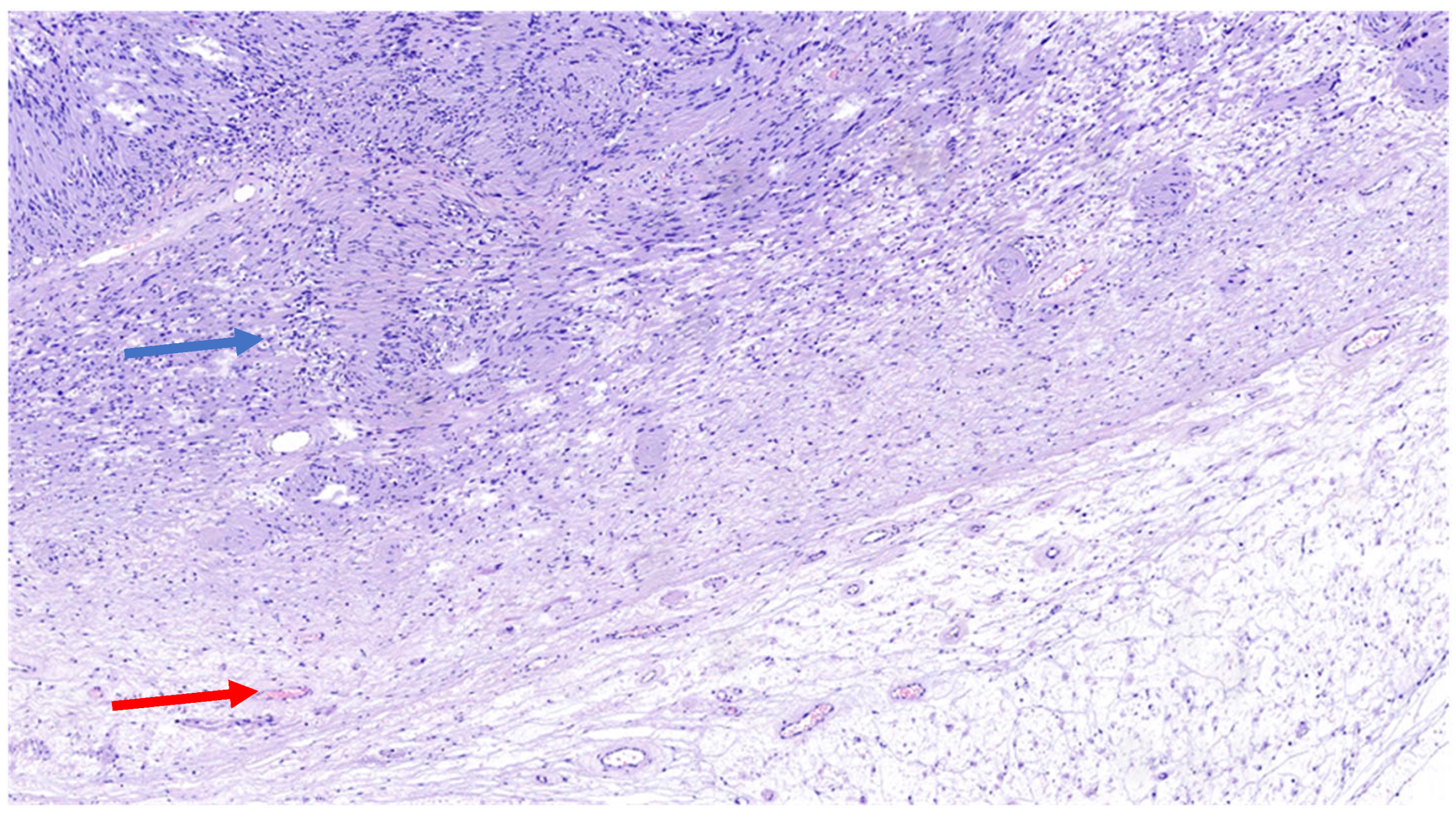
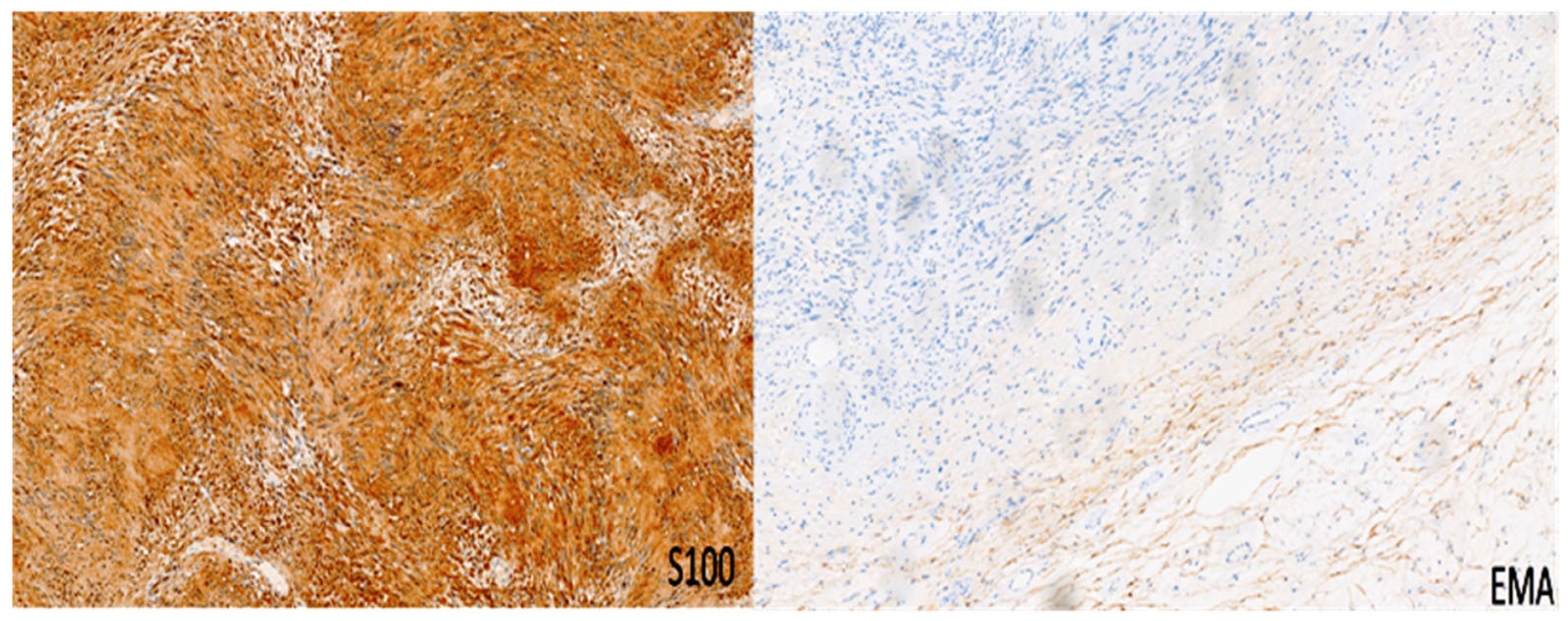
| Schwannoma | All ages; M = F | Limbs | Biphasic: compact hypercellular Antoni A areas and myxoid hypocellular Antoni B areas; nuclear palisading around fibrillary process (Verocay bodies). | S100 + SOX10 + Podoplanin + EMA- | SMARCB1 NF2 | Neurofibromatosis type 2, schwannomatosis, Carney complex |
| Neurofibroma | Second–third decades; M = F | Body surface usually in the head and neck region | Schwann cells with wire-like collagen fibrils, stromal mucosubstances, mast cells, Wagner–Meissner corpuscles, Pacinian corpuscles, axons, fibroblasts, and collagen. | S100 + CD34 + SOX10 + Factor XIIIa + Calretinin + EMA + Podoplanin + | NF1 | Neurofibromatosis type 1 |
| Perineurioma | Young adults; M = F | Limbs | Multiple small “onion bulbs” expanding the affected nerve, consisting of concentric layers of perineurial cells ensheathing a central axon and Schwann cell | EMA + CD34 + S100 + GFAP – CD57 − | TRAF7 NF1 NF2 | No |
| Author | Cases (n) | Gender | Age | Anatomic Site | PNST Type | Follow-Up | Histopathology | Genetic Associations | Tumor Syndromes | Imaging | Local Recurrence | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Agaimy [10] | 2 | 1 female 1 male | 50; 17 | gastric antrum; vermiform appendix | Schwannoma–perineurioma | 8–12 months | storiform, lamellar, fascicular patterns | No | No | N.A. | No | surgery |
| Bergamini [11] | 1 | Female | 54 | mandibular body | Schwannoma–perineurioma | 2 years | spindle to epithelioid cells; wavy and hyperchromatic nuclei. | No | No | N.A. | No | surgery |
| Chow [4] | 1 | Male | 18 | femur | Schwannoma–perineurioma | 7 months | storiform pattern; wavy nuclei | No | No | Plain radiograph: displaced fracture in right femur diaphysis with expansile osteolytic lesion with well-defined borders. | No | surgery |
| Lang [12] | 1 | Female | 28 | tibia | Schwannoma–perineurioma | 8 months | bland-looking spindle to epithelioid cells | No | No | Radiological examination revealed an oval eccentric osteolytic lesion in the proximal tibia. | No | surgery |
| Hornick [13] | 42 | 22 female 20 male | mean age: 38 y; range: 2 to 85 | lower limb (19), upper limb (12), head and neck (6), trunk (4), colon (1) | Schwannoma–perineurioma | 24 months | storiform, whorled/lamellar pattern. Infiltrative margins in 1 case | No | No | N.A. | 1/42 | surgery |
| Singh [14] | 24 | 15 Female 9 Male | mean age: 50 years (range: 24–78 years) | spinal roots (19) intramuscular (5) | Schwannoma--perineurioma | N.A. | dual Schwannian and perineural differentiation. | No | No | MRI: lobular contour, ancient changes, fascicular sign, entering nerve sign, exiting nerve sign, target sign, and split fat sign | No | surgery |
| Ud Din [15] | 5 | 1 female 4 male | mean: 24 years; median: 12 years | big toe of left foot (1), soft tissue of left thigh (1), soft tissue of right thigh (1), soft tissue of neck region (1), retroperitoneum (1) | Schwannoma–perineurioma (3), neurofibroma–perineurioma (1), schwannoma–neurofibroma (1) | N.A. | Spindle cells | No | No | N.A. | No | surgery |
| Michal [3] | 6 | 5 Female 1 Male | mean age: 33 years | fingers (5), thenar eminence of the hand (1) | Schwannoma–retiform perineurioma | N.A. | myxoid and pseudocystic changes. | No | No | N.A. | No | surgery |
| Kuroda [16] | 1 | Male | 58 | middle meatus of the nose | Schwannoma–neurofibroma–perineurioma | N.A. | schwannoma, neurofibroma, and perineurioma differentiation. | No | No | N.A. | No | surgery |
| Chijiiwa [17] | 1 | Male | 41 | right thigh | Schwannoma–neurofibroma | N.A. | dual histology (schwannoma and neurofibroma). | No | No | Well-circumscribed intramuscular mass with low-to-intermediate signal intensity on T1-weighted sequences and higher signal intensity peripherally and lower signal intensity centrally | No | surgery |
| Leite [18] | 1 | Female | 68 | lower vestibule | Neurofibroma–schwannoma | N.A. | dual histology (schwannoma and neurofibroma). | No | No | N.A. | No | surgery |
| Emanuel [19]. | 1 | Male | 48 | colon | Perineurioma–schwannoma | N.A. | whorling or storiform growth pattern mixed with spindle wavy cells | No | No | Colonoscopy and computed tomography scan revealed an obstructing colonic mass, causing intussusception and pneumatosis of the descending/upper sigmoid colon and necessitating an emergency left hemicolectomy | No | surgery |
| Colazo [20] | 1 | Male | 74 | ulnar nerve | Neurofibroma–schwannoma | N.A. | “thumbprint” pattern of neurofibroma; spindled cells arranged in fascicles and palisades | BRAF, TERT and NF2 | No | suspicious for a PNST, but an ultrasound-guided biopsy was equivocal; mimicked glandular schwannoma | No | surgery |
| Goyal-Honavar [21] | 1 | Female | 22 | trigeminal nerve | Schwannoma–perineurioma. | 5 months | biphasic pattern with areas of spindle-shaped cells | No | No | MRI: solid and cystic extra-axial tumor in the right cerebellopontine angle cistern | No | surgery |
| Hong [22] | 1 | Male | 54 | orbit | Neurofibroma–schwannoma | 4 months | spindle cells | No | No | MRI: well-demarcated tumor of 43 mm compressing the optic nerve medially | No | surgery |
| Kacerovska [8] | 4 | N.A. | N.A. | N.A. | Schwannian–perineurioma (3) neurofibroma–perineurioma (1) | N.A. | spindle cells | NF1 | Type 1 neurofibromatosis | N.A. | No | surgery |
| Mitsui [23] | 3 | N.A. | N.A. | upper back, forearm, and thigh | Schwannoma–neurofibroma | N.A. | spindle cells | No | No | N.A. | No | surgery |
| Yusuke Inatomi [24] | 1 | Male | 30 | head | Perineurioma–neurofibroma | 2 months | N.A. | NF1 | Type 1 neurofibromatosis | N.A. | No | surgery |
| Harder [25] | 14 | N.A. | N.A. | N.A. | Neurofibroma–schwannoma | N.A. | N.A. | NF2 | Schwannomatosis | N.A. | No | surgery |
| McLaughlin [26] | 1 | Male | 30 | upper thoracic–region | Schwannoma–perineurioma–neurofibroma | 5 years | schwannoma, neurofibroma, and perineurioma differentiation. | NF1 | Type 1 neurofibromatosis | N.A. | No | surgery |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salzano, S.; Caltabiano, R.; Zanelli, M.; Palicelli, A.; Zizzo, M.; Koufopoulos, N.; Boutas, I.; Magro, G.; Barresi, V.; Broggi, G. Hybrid Benign Peripheral Nerve Sheath Tumors: A Comprehensive Literature Review with Emphasis on Their Clinical, Morphological and Genetic Features. Diagnostics 2025, 15, 855. https://doi.org/10.3390/diagnostics15070855
Salzano S, Caltabiano R, Zanelli M, Palicelli A, Zizzo M, Koufopoulos N, Boutas I, Magro G, Barresi V, Broggi G. Hybrid Benign Peripheral Nerve Sheath Tumors: A Comprehensive Literature Review with Emphasis on Their Clinical, Morphological and Genetic Features. Diagnostics. 2025; 15(7):855. https://doi.org/10.3390/diagnostics15070855
Chicago/Turabian StyleSalzano, Serena, Rosario Caltabiano, Magda Zanelli, Andrea Palicelli, Maurizio Zizzo, Nektarios Koufopoulos, Ioannis Boutas, Gaetano Magro, Valeria Barresi, and Giuseppe Broggi. 2025. "Hybrid Benign Peripheral Nerve Sheath Tumors: A Comprehensive Literature Review with Emphasis on Their Clinical, Morphological and Genetic Features" Diagnostics 15, no. 7: 855. https://doi.org/10.3390/diagnostics15070855
APA StyleSalzano, S., Caltabiano, R., Zanelli, M., Palicelli, A., Zizzo, M., Koufopoulos, N., Boutas, I., Magro, G., Barresi, V., & Broggi, G. (2025). Hybrid Benign Peripheral Nerve Sheath Tumors: A Comprehensive Literature Review with Emphasis on Their Clinical, Morphological and Genetic Features. Diagnostics, 15(7), 855. https://doi.org/10.3390/diagnostics15070855