
Porto-Pulmonary Hypertension and Hepato-Pulmonary Syndrome: Diagnostic Procedures and Therapeutic Management



Abstract
1. Introduction
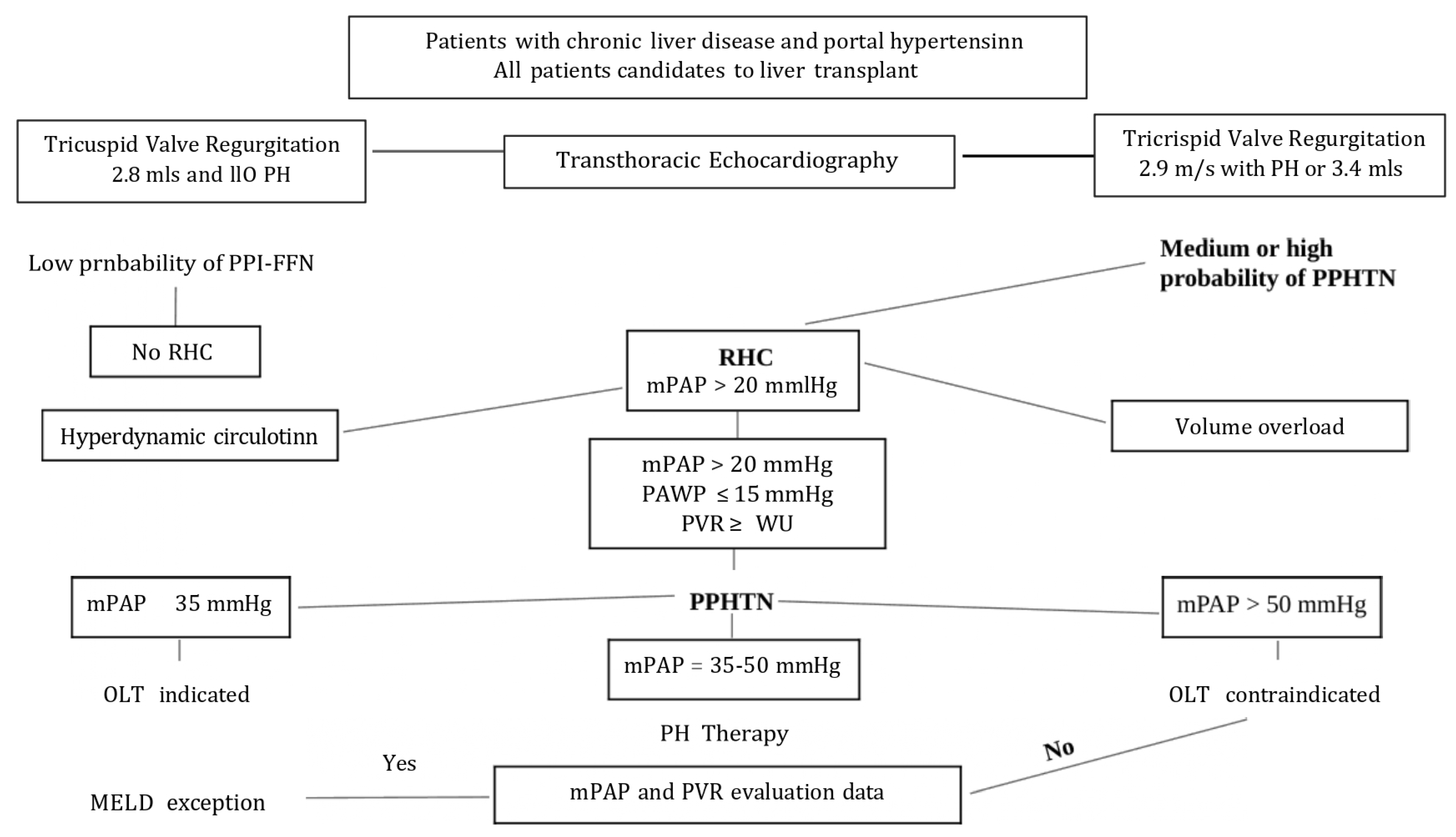
2. Porto-Pulmonary Hypertension
- Mean pulmonary artery pressure (mPAP) > 20 mmHg
- Pulmonary capillary wedge pressure (PCWP) < 15 mmHg
- Pulmonary vascular resistance (PVR) ≥ 2 Wood units (WU)
2.1. Demographics and Characteristics of Patients
2.2. Pathophysiology
2.3. New Insights in Pathophysiology
2.4. Screening and Diagnosis
2.5. Treatment
2.6. Liver Transplant
2.7. Outcome
3. Hepato-Pulmonary Syndrome
3.1. Demographics and Characteristics of Patients
3.2. Pathophysiology
3.3. Screening and Diagnosis
3.4. Treatment
3.4.1. Endothelin Receptor Antagonists
3.4.2. Phosphodiesterase-5 Inhibitors
3.4.3. Soluble Guanylate Cyclase Stimulator
3.4.4. Prostacyclin Analogues
3.4.5. Combination Therapy
3.5. Trans Jugular Intrahepatic Portosystemic Shunt
4. HPS Outcome
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naeije, R. Hepato-pulmonary syndrome and porto-pulmonary hypertension. Swiss Med. Wkly. 2003, 133, 163–169. [Google Scholar]
- Chhabria, M.S.; Boppana, L.K.T.; Manek, G.; Tonelli, A.R. Portopulmonary hypertension: A focused review for the internist. Clevel. Clin. J. Med. 2023, 90, 632–639. [Google Scholar] [CrossRef]
- Aldenkortt, F.; Aldenkortt, M.; Caviezel, L.; Waeber, J.L.; Weber, A.; Schiffer, E. Portopulmonary hypertension and hepatopulmonary syndrome. World J. Gastroenterol. 2014, 20, 8072–8081. [Google Scholar] [CrossRef] [PubMed]
- DuBrock, H.M.; Cartin-Ceba, R.; Channick, R.N.; Kawut, S.M.; Krowka, M.J. Sex differences in portopulmonary hypertension. Chest 2021, 159, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef]
- Krowka, M.J.; Fallon, M.B.; Kawut, S.M.; Fuhrmann, V.; Heimbach, J.K.; Ramsay, M.A.E.; Sitbon, O.; Sokol, R.J. International Liver Transplant Society Practice Guidelines: Diagnosis and Management of Hepatopulmonary Syndrome and Portopulmonary Hypertension. Transplantation 2016, 100, 1440–1452. [Google Scholar] [CrossRef]
- De Franchis, R. and Baveno VI Faculty. Expanding consensus in portal hypertension. Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J. Hepatol. 2015, 63, 743–752. [Google Scholar] [CrossRef]
- McDonnell, P.J.; Toye, P.A.; Hutchins, G.M. Primary pulmonary hypertension and cirrhosis: Are they related? Am. Rev. Respir. Dis. 1983, 127, 437–441. [Google Scholar] [CrossRef]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef]
- Savale, L.; Guimas, M.; Ebstein, N.; Fertin, M.; Jevnikar, M.; Renard, S.; Horeau-Langlard, D.; Tromeur, C.; Chabanne, C.; Prevot, G.; et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J. Hepatol. 2020, 73, 130–139. [Google Scholar] [CrossRef]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Krowka, M.J.; Miller, D.P.; Barst, R.J.; Taichman, D.; Dweik, R.A.; Badesch, D.B.; McGoon, M.D. Portopulmonary hypertension. A report from the US-based Reveal Registry. Chest 2012, 141, 906–915. [Google Scholar] [CrossRef]
- Hadengue, A.; Benhayoun, M.K.; Lebrec, D.; Benhamou, J.P. Pulmonary hypertension complicating portal hypertension: Prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991, 100, 520–528. [Google Scholar] [CrossRef]
- Bolognesi, M.; Di Pascoli, M.; Verardo, A.; Gatta, A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J. Gastroenterol. 2014, 20, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Al-Naamani, N.; Krowka, M.J.; Forde, K.A.; Krok, K.L.; Feng, R.; Heresi, G.A.; Dweik, R.A.; Bartolome, S.; Bull, T.M.; Roberts, K.E.; et al. Estrogen signaling and portopulmonary hypertension: The pulmonary vascular complications of liver disease study (PVCLD2). Hepatology 2021, 73, 726–737. [Google Scholar] [CrossRef]
- Edwards, B.S.; Weir, E.K.; Edwards, W.D.; Ludwig, J.; Dykoski, R.K.; Edwards, J.E. Coexistent pulmonary and portal hypertension: Morphologic and clinical features. J. Am. Coll. Cardiol. 1987, 10, 1233–1238. [Google Scholar] [CrossRef]
- Kamm, D.R.; McCommis, K.S. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Benz, F.; Mohr, R.; Tacke, F.; Roderburg, C. Pulmonary complications in patients with liver cirrhosis. J. Transl. Intern. Med. 2020, 8, 150–158. [Google Scholar] [CrossRef]
- Thomas, C.; Glinskii, V.; de Jesus Perez, V.; Sahay, S. Portopulmonary hypertension: From bench to bedside. Front. Med. 2020, 7, 569413. [Google Scholar] [CrossRef]
- Tokushige, K.; Kogiso, T.; Egawa, H. Current therapy and liver transplantation for portopulmonary hypertension in Japan. J. Clin. Med. 2023, 12, 562. [Google Scholar] [CrossRef]
- DuBrock, H.M. Portopulmonary Hypertension. Management and liver transplantation evaluation. Chest 2023, 164, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Jasso-Baltazar, E.A.; Peña-Arellano, G.A.; Aguirre-Valadez, J.; Ruiz, I.; Papacristofilou-Riebeling, B.; Jimenez, J.V.; García-Carrera, C.J.; Rivera-López, F.E.; Rodriguez-Andoney, J.; Lima-Lopez, F.C.; et al. Portopulmonary hypertension: An updated review. Transplant. Direct 2023, 9, e1517. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, B.; Wang, R.; Ding, M.; Gao, Y. Portopulmonary hypertension: Current developments and future perspectives. Liver Res. 2022, 6, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Deroo, R.; Trépo, E.; Holvoet, T.; De Pauw, M.; Geerts, A.; Verhelst, X.; Colle, I.; Van Vlierberghe, H.; Fallon, M.B.; Raevens, S. Vasomodulators and liver transplantation for portopulmonary hypertension: Evidence from a systematic review and meta-analysis. Hepatology 2020, 72, 1701–1716. [Google Scholar] [CrossRef]
- Tamura, Y.; Tamura, Y.; Taniguchi, Y.; Atsukawa, M. Current clinical understanding and effectiveness of portopulmonary hypertension treatment. Front. Med. 2023, 10, 1142836. [Google Scholar] [CrossRef]
- Cartin-Ceba, R.; Burger, C.; Swanson, K.; Vargas, H.; Aqel, B.; Keaveny, A.P.; Heimbach, J.; Taner, T.; Nyberg, S.; Rosen, C.; et al. Clinical outcomes after liver transplantation in patients with portopulmonary hypertension. Transplantation 2021, 105, 2283–2290. [Google Scholar] [CrossRef]
- Peppas, S.; Nagraj, S.; Koutsias, G.; Kladas, M.; Archontakis-Barakakis, P.; Schizas, D.; Giannakoulas, G.; Palaiodimos, L.; Kokkinidis, D.G. Portopulmonary hypertension: A review of the current literature. Heart Lung Circ. 2022, 31, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Bosch, J.; Cottreel, E.; Csonka, D.; De Groote, P.; Hoeper, M.M.; Kim, N.H.; Martin, N.; Savale, L.; Krowka, M. Macitentan for the treatment of portopulmonary hypertension (PORTICO): A multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir. Med. 2019, 7, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Angermayr, B.; Cejna, M.; Karnel, F.; Gschwantler, M.; Koenig, F.; Pidlich, J.; Mendel, H.; Pichler, L.; Wichlas, M.; Kreil, A.; et al. Child-Pugh versus MELD score in predicting survival in patients undergoing transjugular intrahepatic portosystemic shunt. Gut 2003, 52, 879–885. [Google Scholar] [CrossRef] [PubMed]
- AbuHalimeh, B.; Krowka, M.J.; Tonelli, A.R. Treatment barriers in portopulmonary hypertension. Hepatology 2019, 69, 431–443. [Google Scholar] [CrossRef]
- Krowka, M.; Plevak, D.J.; Findlay, J.Y.; Rosen, C.B.; Wiesner, R.H.; Krom, R.A. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000, 6, 443–450. [Google Scholar] [CrossRef]
- Giannini, E.G.; Savarino, V.; Farinati, F.; Ciccarese, F.; Rapaccini, G.; Di Marco, M.; Benvegnù, L.; Zoli, M.; Borzio, F.; Caturelli, E.; et al. Influence of clinically significant portal hypertension on survival after hepatic resection for hepatocellular carcinoma in cirrhotic patients. Liver Int. 2013, 33, 1594–1600. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Valapour, M.; Skeans, M.A.; Heubner, B.M.; Smith, J.M.; Schnitzler, M.A.; Hertz, M.I.; Edwards, L.B.; Snyder, J.J.; Israni, A.K.; Kasiske, B.L. OPTN/SRTR 2012 annual data report: Lung. Am. J. Transplant. 2014, 14 (Suppl. 1), 139–165. [Google Scholar] [CrossRef]
- Schenk, P.; Fuhrmann, V.; Madl, C.; Funk, G.; Lehr, S.; Kandel, O.; Müller, C. Hepatopulmonary syndrome: Prevalence and predictive value of various cut offs for arterial oxygenation and their clinical consequences. Gut 2002, 51, 853–859. [Google Scholar] [CrossRef]
- Stoller, J.K.; Lange, P.A.; Westveer, M.K.; Carey, W.D.; Vogt, D.; Henderson, J.M. Prevalence and reversibility of the hepatopulmonary syndrome after liver transplantation. The Cleveland Clinic experience. West J. Med. 1995, 163, 133–138. [Google Scholar]
- Kim, B.J.; Lee, S.C.; Park, S.W.; Choi, M.S.; Koh, K.C.; Paik, S.W.; Choi, M.S.; Koh, K.C.; Paik, S.W.; Lee, S.H.; et al. Characteristics and prevalence of intrapulmonary shunt detected by contrast echocardiography with harmonic imaging in liver transplant candidates. Am. J. Cardiol. 2004, 94, 525–528. [Google Scholar] [CrossRef]
- Rodriguez-Roisin, R.; Krowka, M.J.; Hervé, P.; Fallon, M.B.; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-Hepatic vascular Disorders (PHD). Eur. Respir. J. 2004, 24, 861–880. [Google Scholar] [CrossRef]
- Rodríguez-Roisin, R.; Krowka, M.J. Hepatopulmonary syndrome—A liver-induced lung vascular disorder. N. Engl. J. Med. 2008, 358, 2378–2387. [Google Scholar] [CrossRef]
- Dzikowska-Diduch, O.; Cader, T.; Jankowski, K.; Ou-Pokrzewinska, A.; Sznajder, M.; Siwiec, J.; Pucyło, S.; Sikora, A.; Pacholczyk, M.; Lisik, W.; et al. Echocardiographic screening of liver transplant candidates—Prevalence of features of portopulmonary hypertension. J. Clin. Med. 2024, 1, 6990. [Google Scholar] [CrossRef]
- Krowka, M.J. Portopulmonary hypertension. Semin. Respir. Crit. Care Med. 2012, 33, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Magnier, R.; Le Pavec, J.; Jais, X.; Montani, D.; O’Callaghan, D.S.; Humbert, M.; Dingemanse, J.; Simonneau, G.; Sitbon, O. Efficacy, safety, and pharmacokinetics of bosentan in portopulmonary hypertension. Eur. Res. J. 2013, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Seyfarth, H.J.; Hoeffken, G.; Wirtz, H.; Spiekerkoetter, E.; Pletz, M.W.; Welte, T.; Halank, M. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur. Respir. J. 2007, 30, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Halank, M.; Marx, C.; Hoeffken, G.; Seyfarth, H.J.; Schauer, J.; Niedermeyer, J.; Winkler, J. Bosentan therapy for portopulmonary hypertension. Eur. Respir. J. 2005, 25, 502–508. [Google Scholar] [CrossRef]
- Preston, I.R.; Burger, C.D.; Bartolome, S.; Safdar, Z.; Krowka, M.; Sood, N.; Ford, H.J.; Battarjee, W.F.; Chakinala, M.M.; Gomberg-Maitland, M.; et al. Ambrisentan in portopulmonary hypertension: A multicenter, open-label trial. J. Heart Lung Transpl. 2020, 39, 464–472. [Google Scholar] [CrossRef]
- Fisher, J.H.; Johnson, S.R.; Chau, C.; Kron, A.T.; Granton, J.T. Effectiveness of phosphodiesterase-5 inhibitor therapy for portopulmonary hypertension. Can. Respir. J. 2015, 22, 42–46. [Google Scholar] [CrossRef]
- Reichenberger, F.; Voswinckel, R.; Steveling, E.; Enke, B.; Kreckel, A.; Olschewski, H.; Grimminger, F.; Seeger, W.; Ghofrani, A. Sildenafil treatment for portopulmonary hypertension. Eur. Respir. J. 2006, 28, 563–567. [Google Scholar] [CrossRef]
- Gough, M.S.; White, R.J. Sildenafil therapy is associated with improved hemodynamics in liver transplantation candidates with pulmonary arterial hypertension. Liver Transpl. 2009, 15, 30–36. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Robbins, I.M. Sildenafil monotherapy in portopulmonary hypertension can facilitate liver transplantation. Liver Transpl. 2009, 15, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Talarico, M.; Schepis, F.; Coppi, F.; Sgura, F.A.; Monopoli, D.E.; Minici, R.; Boriani, G. Effects of sildenafil on right ventricular remodeling in portopulmonary hypertension. Pulm. Pharmacol. Ther. 2021, 70, 102071. [Google Scholar] [CrossRef] [PubMed]
- Sussman, N.; Kaza, V.; Barshes, N.; Stribling, R.; Goss, J.; O’Mahony, C.; Zhang, E.; Vierling, J.; Frost, A. Successful liver transplantation following medical management of portopulmonary hypertension: A single-centre series. Am. J. Transpl. 2006, 6, 2177–2182. [Google Scholar] [CrossRef] [PubMed]
- Melgosa, M.T.; Ricci, G.L.; García-Pagan, J.C.; Blanco, I.; Escribano, P.; Abraldes, J.G.; Roca, J.; Bosch, J.; Barberà, J.A. Acute and long-term effects of inhaled iloprost in portopulmonary hypertension. Liver Transpl. 2010, 16, 348–356. [Google Scholar] [CrossRef]
- Tamura, Y.; Tamura, Y.; Taniguchi, Y.; Tsujino, I.; Inami, T.; Matsubara, H.; Shigeta, A.; Sugiyama, Y.; Adachi, S.; Abe, K.; et al. Clinical management and outcomes of patients with portopulmonary hypertension enrolled in the Japanese multicentre registry. Circ. Rep. 2022, 4, 542–549. [Google Scholar] [CrossRef]
- Boyer, T.D.; Haskal, Z.J. The role of transjugular intrahepatic portosystemic shunt (TIPS) in the management of portal hypertension: Update 2009. Hepatology 2010, 51, 306. [Google Scholar] [CrossRef]
- Bansal, K.; Gore, M.; Mittal, S. Hepatopulmonary Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porto-Pulmonary Hypertension | Hepato-Pulmonary Syndrome |
|---|---|
| Group 1 PAH clinical classification | Liver disease (usually cirrhosis with portal hypertension) |
| PAH in patients with portal hypertension | Positive contrast-enhanced transthoracic echocardiography |
| Elevated mean pulmonary arterial pressure (mPAP) | Hypoxemia: alveolar-arterial gradient (A-a O2) ≥15 mmHg (or ≥20 if >65 years) Hypoxemia: O2 arterial partial pressure (PaO2) <70 mmHg |
| Increased pulmonary vascular resistance (PVR) | |
| Right-sided heart failure | Intrapulmonary vascular dilatations and/or shunting |
| High morbidity and mortality |
| Child-Turcotte-Pugh Classification | |||
|---|---|---|---|
| Score | 1 | 2 | 3 |
| Ascites | Absent | Moderate | Massive |
| Encephalopathy | Absent | Mild | Severe |
| Bilirubin (mg/dL) | <2 | 2–3 | >3 |
| Albumin (g/dL) | >3.5 | 2.8–3.5 | <2.8 |
| Prothrombin time (%) | >70 | 40–70 | <40 |
| Authors | No. of Patients | Survival (%) | ||
|---|---|---|---|---|
| 1 yr | 3 yrs | 5 yrs | ||
| Sadd et al. | 24 | 87 | 87 | 87 |
| Reymond et al. | 23 | 83 | 83 | -- |
| Rajaram et al. | 13 | 69 | -- | -- |
| Ashfaq et al. | 11 | 91 | -- | 67 |
| Cartin-Ceba et al. | 50 | 72 | 63 | 60 |
| Salgia et al. | 78 | 85 | 81 | -- |
| Verma et al. | 28 | 63 | 59 | 54 |
| DuBrock et al. | 103 | 86 | – | -- |
| Savale et al. | 35 | 80 | 77 | 77 |
| Savale et al. | 63 | 92 | 83 | 81 |
| Differential Diagnosis | Diagnostic Procedures |
|---|---|
| Porto-pulmonary Hypertension | Contrast-enhanced echocardiography transesophageal echocardiogram |
| Atelectasis | Chest computed tomography |
| Recurrent pulmonary emboli | Pulmonary angiography |
| Atrial septal defect | Transesophageal echocardiogram |
| Arteriovenous malformations | Pulmonary angiography |
| Post-pneumonectomy | Echocardiography |
| Chronic cardiopulmonary disease | Chest computed tomography Pulmonary function tests |
| COPD | Pulmonary function tests Echocardiography |
| Pneumonitis | Chest X-ray |
| Hepatic hydrothorax | Total body computed tomography Magnetic resonance imaging |
| Ascites | Abdominal ultrasonogram |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, R.G.; Puppo, F.; Thomas, C.A.; Savarino, V. Porto-Pulmonary Hypertension and Hepato-Pulmonary Syndrome: Diagnostic Procedures and Therapeutic Management. Diagnostics 2025, 15, 1821. https://doi.org/10.3390/diagnostics15141821
Carbone RG, Puppo F, Thomas CA, Savarino V. Porto-Pulmonary Hypertension and Hepato-Pulmonary Syndrome: Diagnostic Procedures and Therapeutic Management. Diagnostics. 2025; 15(14):1821. https://doi.org/10.3390/diagnostics15141821
Chicago/Turabian StyleCarbone, Roberto G., Francesco Puppo, Christopher A. Thomas, and Vincenzo Savarino. 2025. "Porto-Pulmonary Hypertension and Hepato-Pulmonary Syndrome: Diagnostic Procedures and Therapeutic Management" Diagnostics 15, no. 14: 1821. https://doi.org/10.3390/diagnostics15141821
APA StyleCarbone, R. G., Puppo, F., Thomas, C. A., & Savarino, V. (2025). Porto-Pulmonary Hypertension and Hepato-Pulmonary Syndrome: Diagnostic Procedures and Therapeutic Management. Diagnostics, 15(14), 1821. https://doi.org/10.3390/diagnostics15141821