
Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Study Population
2.3. Data Collection and Procedures
2.4. Statistical Analysis
3. Results
3.1. Baseline Characteristics of the Study Group
3.2. BALF Cell Analysis
3.3. Predictors of Fibrotic HP Diagnosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernández Pérez, E.R.; Travis, W.D.; Lynch, D.A.; Brown, K.K.; Johannson, K.A.; Selman, M.; Ryu, J.H.; Wells, A.U.; Tony Huang, Y.-C.; Pereira, C.A.C.; et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest 2021, 160, e97–e156. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef] [PubMed]
- Lacasse, Y.; Selman, M.; Costabel, U.; Dalphin, J.-C.; Ando, M.; Morell, F.; Erkinjuntti-Pekkanen, R.; Muller, N.; Colby, T.V.; Schuyler, M.; et al. Clinical diagnosis of hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2003, 168, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Morell, F.; Villar, A.; Montero, M.-Á.; Muñoz, X.; Colby, T.V.; Pipvath, S.; Cruz, M.-J.; Raghu, G. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: A prospective case-cohort study. Lancet Respir. Med. 2013, 1, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Vasakova, M.; Morell, F.; Walsh, S.; Leslie, K.; Raghu, G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2017, 196, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Walsh, S.L.F.; Wells, A.U.; Desai, S.R.; Poletti, V.; Piciucchi, S.; Dubini, A.; Nunes, H.; Valeyre, D.; Brillet, P.Y.; Kambouchner, M.; et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: A case-cohort study. Lancet Respir. Med. 2016, 4, 557–565. [Google Scholar] [CrossRef]
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef]
- Reynolds, H.Y. Bronchoalveolar lavage and other methods to define the human respiratory tract milieu in health and disease. Lung 2011, 189, 87–99. [Google Scholar] [CrossRef]
- Caillaud, D.M.; Vergnon, J.M.; Madroszyk, A.; Melloni, B.M.; Murris, M.; Dalphin, J.C. French Group of Environmental Immunoallergic Bronchopulmonary Diseases Bronchoalveolar lavage in hypersensitivity pneumonitis: A series of 139 patients. Inflamm. Allergy-Drug Targets 2012, 11, 15–19. [Google Scholar] [CrossRef]
- Tzilas, V.; Tzouvelekis, A.; Bouros, E.; Karampitsakos, T.; Ntasiou, M.; Katsaras, M.; Costabel, U.; Wells, A.; Bouros, D. Diagnostic value of BAL lymphocytosis in patients with indeterminate for usual interstitial pneumonia imaging pattern. Eur. Respir. J. 2019, 54, 1901144. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Barańska, I.; Jędrych, M.E.; Bartoszuk, I.; Radwan-Roehrenschef, P.; Roży, A.; Bestry, I.; Chorostowska-Wynimko, J.; Langfort, R.; Kuś, J. Hypersensitivity pneumonitis recognised in a single pulmonary unit, between 2005 and 2015—Comparison with recently proposed diagnostic criteria. Adv. Respir. Med. 2019, 87, 83–89. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Barańska, I.; Skoczylas, A.; Jędrych, M.E.; Demkow, U. Correlation of bronchoalveolar lavage lymphocyte count with the extent of lung fibrosis and with plethysmographic lung volumes in patients with newly recognized hypersensitivity pneumonitis. Cent.-Eur. J. Immunol. 2020, 45, 276–282. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef]
- Wanger, J.; Clausen, J.L.; Coates, A.; Pedersen, O.F.; Brusasco, V.; Burgos, F.; Casaburi, R.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 2005, 26, 511–522. [Google Scholar] [CrossRef]
- Quanjer, P.H.; Tammeling, G.J.; Cotes, J.E.; Pedersen, O.F.; Peslin, R.; Yernault, J.C. Lung volumes and forced ventilatory flows. Eur. Respir. J. 1993, 6 (Suppl. 16), 5–40. [Google Scholar] [CrossRef]
- Graham, B.L.; Brusasco, V.; Burgos, F.; Cooper, B.G.; Jensen, R.; Kendrick, A.; MacIntyre, N.R.; Thompson, B.R.; Wanger, J. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur. Respir. J. 2017, 49, 1600016. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: Guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef]
- Liu, X. Classification accuracy and cut point selection. Stat. Med. 2012, 31, 2676–2686. [Google Scholar] [CrossRef]
- Chiba, S.; Tsuchiya, K.; Akashi, T.; Ishizuka, M.; Okamoto, T.; Furusawa, H.; Tateishi, T.; Kishino, M.; Miyazaki, Y.; Tateishi, U.; et al. Chronic Hypersensitivity Pneumonitis With a Usual Interstitial Pneumonia-Like Pattern: Correlation Between Histopathologic and Clinical Findings. Chest 2016, 149, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Morisset, J.; Johannson, K.A.; Vittinghoff, E.; Aravena, C.; Elicker, B.M.; Jones, K.D.; Fell, C.D.; Manganas, H.; Dubé, B.-P.; Wolters, P.J.; et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017, 151, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, K.B.; Barańska, I.; Sobiecka, M.; Radwan-Rohrenschef, P.; Dybowska, M.; Franczuk, M.; Roży, A.; Skoczylas, A.; Bestry, I.; Kuś, J.; et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis-Retrospective Cohort Analysis. Diagnostics 2022, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, W.J.; Bestry, I.; Białas, A.J.; Boros, P.W.; Grzanka, P.; Jassem, E.; Jastrzębski, D.; Klimczak, D.; Langfort, R.; Lewandowska, K.; et al. Guidelines of the Polish Respiratory Society for diagnosis and treatment of idiopathic pulmonary fibrosis. Adv. Respir. Med. 2020, 88, 41–93. [Google Scholar] [CrossRef]
- Adderley, N.; Humphreys, C.J.; Barnes, H.; Ley, B.; Premji, Z.A.; Johannson, K.A. Bronchoalveolar lavage fluid lymphocytosis in chronic hypersensitivity pneumonitis: A systematic review and meta-analysis. Eur. Respir. J. 2020, 56, 2000206. [Google Scholar] [CrossRef]
- Patolia, S.; Tamae Kakazu, M.; Chami, H.A.; Chua, A.; Diaz-Mendoza, J.; Duggal, A.; Jenkins, A.R.; Knight, S.L.; Raghu, G.; Wilson, K.C. Bronchoalveolar Lavage Lymphocytes in the Diagnosis of Hypersensitivity Pneumonitis among Patients with Interstitial Lung Disease. Ann. Am. Thorac. Soc. 2020, 17, 1455–1467. [Google Scholar] [CrossRef]
- Morisset, J.; Johannson, K.A.; Jones, K.D.; Wolters, P.J.; Collard, H.R.; Walsh, S.L.F.; Ley, B. HP Delphi Collaborators Identification of Diagnostic Criteria for Chronic Hypersensitivity Pneumonitis: An International Modified Delphi Survey. Am. J. Respir. Crit. Care Med. 2018, 197, 1036–1044. [Google Scholar] [CrossRef]
- Nagai, S.; Handa, T.; Ito, Y.; Takeuchi, M.; Izumi, T. Bronchoalveolar lavage in idiopathic interstitial lung diseases. Semin. Respir. Crit. Care Med. 2007, 28, 496–503. [Google Scholar] [CrossRef]
- Ryu, Y.J.; Chung, M.P.; Han, J.; Kim, T.S.; Lee, K.S.; Chun, E.-M.; Kyung, S.Y.; Jeong, S.H.; Colby, T.V.; Kim, H.; et al. Bronchoalveolar lavage in fibrotic idiopathic interstitial pneumonias. Respir. Med. 2007, 101, 655–660. [Google Scholar] [CrossRef]
- Ohshimo, S.; Bonella, F.; Cui, A.; Beume, M.; Kohno, N.; Guzman, J.; Costabel, U. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 1043–1047. [Google Scholar] [CrossRef]
- Domagała-Kulawik, J.; Skirecki, T.; Maskey-Warzechowska, M.; Grubek-Jaworska, H.; Chazan, R. Bronchoalveolar lavage total cell count in interstitial lung diseases—Does it matter? Inflammation 2012, 35, 803–809. [Google Scholar] [CrossRef]
- Bergantini, L.; d’Alessandro, M.; Cameli, P.; Perrone, A.; Cekorja, B.; Boncompagni, B.; Mazzei, M.A.; Sestini, P.; Bargagli, E. Integrated approach to bronchoalveolar lavage cytology to distinguish interstitial lung diseases. Eur. J. Intern. Med. 2021, 89, 76–80. [Google Scholar] [CrossRef]
- Salisbury, M.L.; Myers, J.L.; Belloli, E.A.; Kazerooni, E.A.; Martinez, F.J.; Flaherty, K.R. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Parameter | fHP | IPF | p-Value |
|---|---|---|---|
| Subjects | 65 | 71 | |
| Age at diagnosis, years | 54.97 (10.87) | 64.00 (7.18) | <0.001 |
| Sex | 0.012 | ||
| Male | 32 (49.2) | 50 (70.4) | |
| Female | 33 (50.8) | 21 (29.6) | |
| Smoking status | <0.001 | ||
| Smokers and ex-smokers | 26 (40.0) | 55 (77.5) | |
| Never-smokers | 39 (60.0) | 16 (22.5) | |
| Pack-years | 8.75 (16.68) | 35.07 (22.08) | <0.001 |
| Identified exposure to: | 47 (72.3) | 8 (11.3) | <0.001 |
| Poultry | 14 | 2 | |
| Pigeons | 16 | 1 | |
| Parrots | 4 | 4 | |
| Hay/feed | 15 | 1 | |
| Other | 10 | 0 |
| Parameter | fHP n = 65 | IPF n = 71 | p-Value |
|---|---|---|---|
| FVC, % predicted | 77.85 (19.29) | 87.14 (17.69) | 0.004 |
| FEV1, %predicted | 73.92 (19.12) | 89.55 (17.02) | <0.001 |
| FEV1/FVC | 80.66 (8.36) | 80.80 (8.85) | 0.924 |
| TLC, %predicted | 79.67 (15.81) | 84.56 (16.85) | 0.088 |
| TL,co, %predicted | 47.20 (16.25) | 52.29 (16.00) | 0.071 |
| 6MWD, m | 496.00 (92.67) | 465.94 (111.12) | 0.092 |
| Desaturation during 6MWT, % | 7.83 (5.91) | 6.38 (5.89) | 0.157 |
| TVPG, mmHg | 31.02 (11.22) | 31.45 (7.72) | 0.815 |
| Parameter | fHP n = 65 | IPF n = 71 | p-Value |
|---|---|---|---|
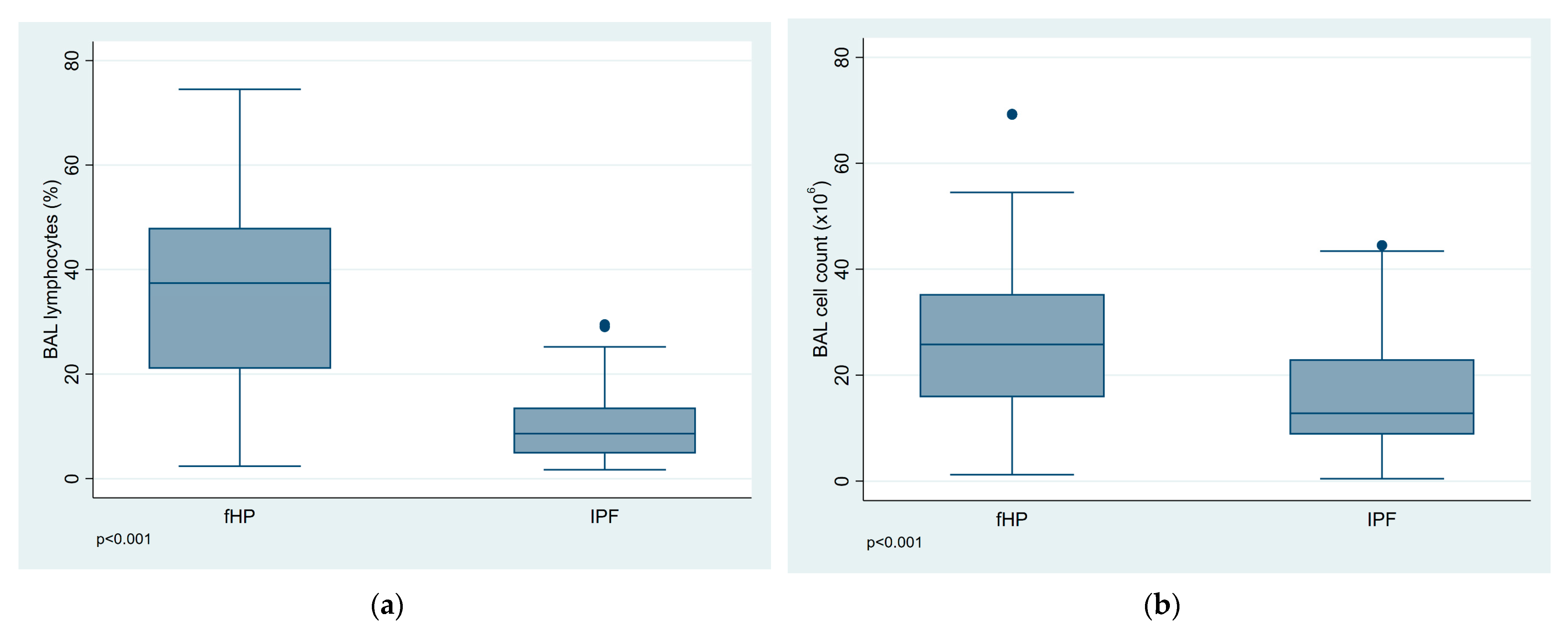
| Total cell count (×106/mL) | 26.23 (15.25) | 16.20 (10.38) | <0.001 |
| Lymphocytes (%) | 35.24 (17.50) | 10.17 (6.80) | <0.001 |
| Eosinophils (%) | 3.11 (3.68) | 3.81 (4.30) | 0.323 |
| Neutrophils (%) | 7.73 (6.93) | 6.74 (6.11) | 0.379 |
| Macrophages (%) | 54.51 (17.47) | 79.28 (10.01) | <0.001 |
| Lymphocytosis ≥ 20%, N (%) | 49 (75.4) | 7 (9.9) | <0.001 |
| BALF lymphocytosis, N (%) | |||
| <10% | 8 (12) | 44 (62) | - |
| 10–20% | 8 (12) | 21 (30) | - |
| 21–30% | 10 (15) | 6 (8) | - |
| 31–40% | 12 (18) | 0 | - |
| 41–50% | 15 (23) | 0 | - |
| 51–60% | 7 (11) | 0 | - |
| >60% | 5 (8) | 0 | - |
| Characteristics | OR | 95% CI | p-Value |
|---|---|---|---|
| Male | 0.90 | 0.36–2.22 | 0.814 |
| Age at diagnosis | 0.90 | 0.86–0.94 | <0.001 |
| Ever smoker | 0.20 | 0.08–0.49 | <0.001 |
| Identified exposure | 17.21 | 5.93–50.00 | <0.001 |
| FVC, % predicted | 0.99 | 0.97–1.01 | 0.424 |
| FEV1, %predicted | 0.97 | 0.95–0.99 | 0.018 |
| FEV1/FVC | 0.97 | 0.93–1.02 | 0.239 |
| TLC, %predicted | 0.99 | 0.96–1.02 | 0.423 |
| TL,co, %predicted | 0.97 | 0.95–1.00 | 0.068 |
| 6MWD, m | 1.00 | 1.00–1.00 | 0.885 |
| Desaturation during 6MWT, % | 1.04 | 0.97–1.12 | 0.237 |
| Total cell count in BALF | 1.04 | 1.01–1.08 | 0.020 |
| Neutrophils in BALF | 1.03 | 0.97–1.10 | 0.340 |
| Eosinophils in BALF | 0.98 | 0.89–1.08 | 0.692 |
| Lymphocytes in BALF | 1.16 | 1.09–1.23 | <0.001 |
| Lymphocytosis in BALF > 20% | 25.06 | 7.43–84.50 | <0.001 |
| Parameter | Cut-off | Specificity | Sensitivity | PPV | NPV | AUC |
|---|---|---|---|---|---|---|
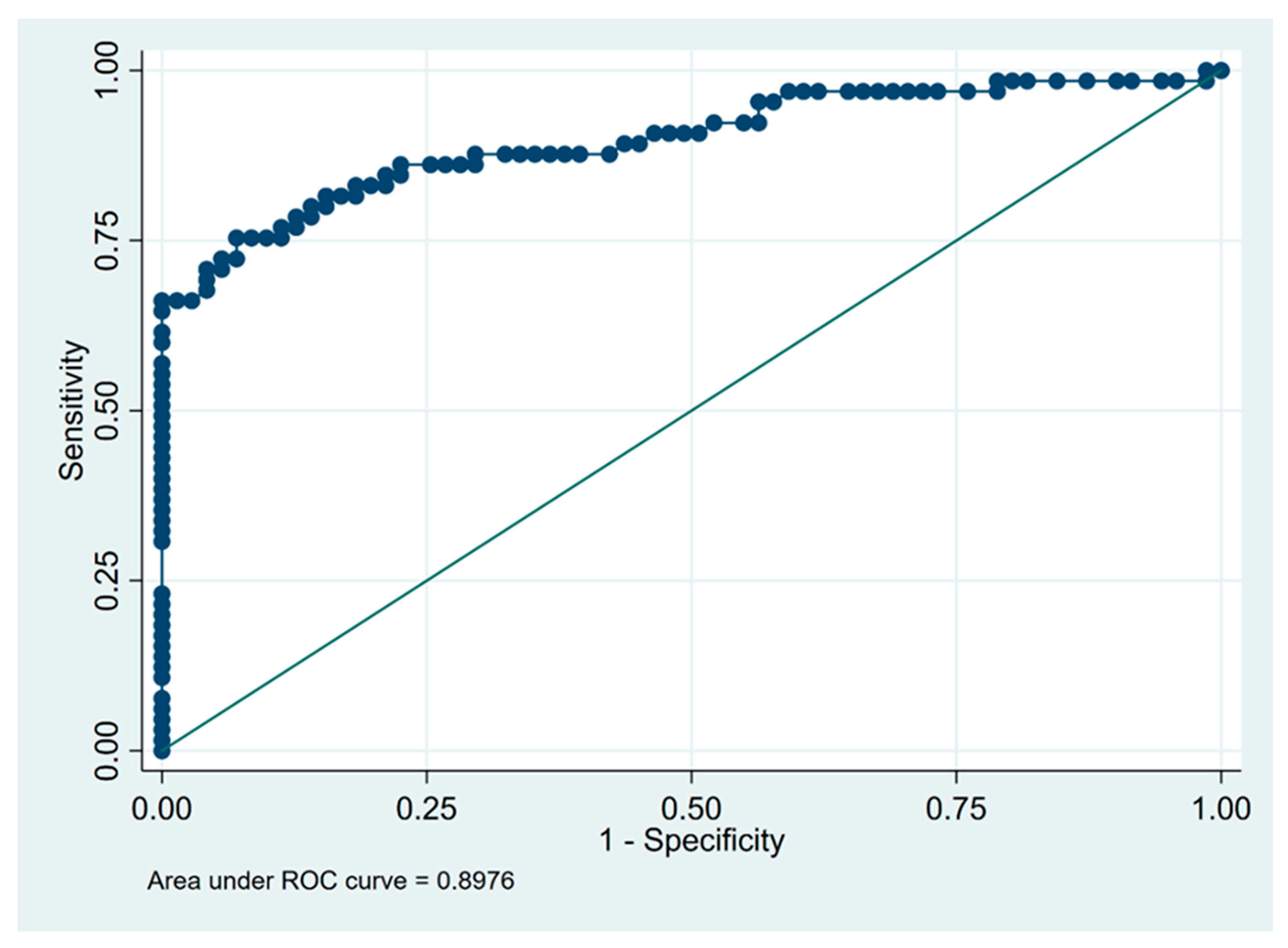
| Lymphocytosis in BALF (%) | 21 | 0.93 | 0.75 | 0.91 | 0.80 | 0.84 |
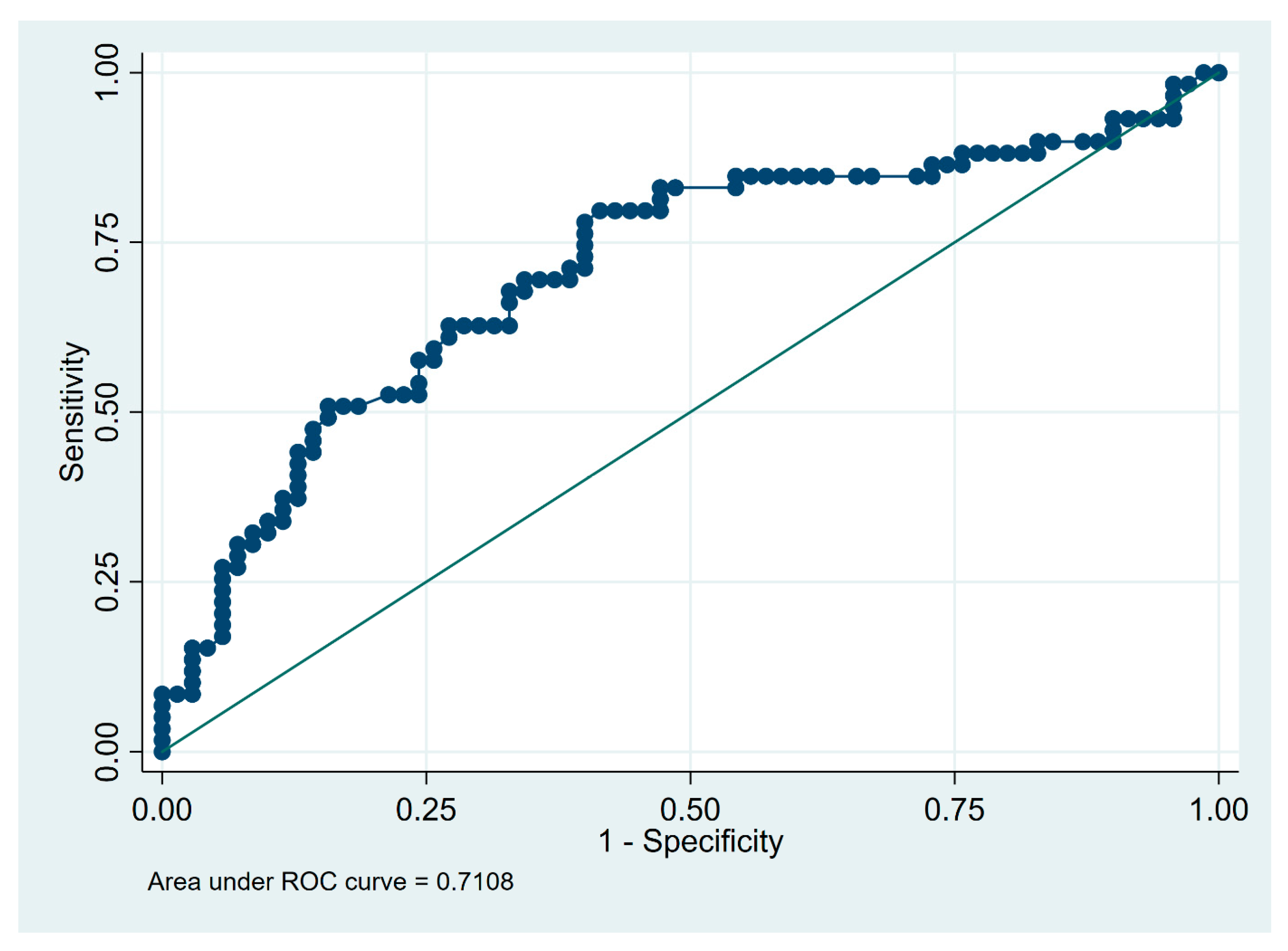
| Total cell count in BALF (×106) | 15 | 0.59 | 0.80 | 0.62 | 0.77 | 0.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobiecka, M.; Szturmowicz, M.; Lewandowska, K.B.; Barańska, I.; Zimna, K.; Łyżwa, E.; Dybowska, M.; Langfort, R.; Radwan-Röhrenschef, P.; Roży, A.; et al. Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis. Diagnostics 2023, 13, 935. https://doi.org/10.3390/diagnostics13050935
Sobiecka M, Szturmowicz M, Lewandowska KB, Barańska I, Zimna K, Łyżwa E, Dybowska M, Langfort R, Radwan-Röhrenschef P, Roży A, et al. Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis. Diagnostics. 2023; 13(5):935. https://doi.org/10.3390/diagnostics13050935
Chicago/Turabian StyleSobiecka, Małgorzata, Monika Szturmowicz, Katarzyna B. Lewandowska, Inga Barańska, Katarzyna Zimna, Ewa Łyżwa, Małgorzata Dybowska, Renata Langfort, Piotr Radwan-Röhrenschef, Adriana Roży, and et al. 2023. "Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis" Diagnostics 13, no. 5: 935. https://doi.org/10.3390/diagnostics13050935
APA StyleSobiecka, M., Szturmowicz, M., Lewandowska, K. B., Barańska, I., Zimna, K., Łyżwa, E., Dybowska, M., Langfort, R., Radwan-Röhrenschef, P., Roży, A., & Tomkowski, W. Z. (2023). Bronchoalveolar Lavage Cell Count and Lymphocytosis Are the Important Discriminators between Fibrotic Hypersensitivity Pneumonitis and Idiopathic Pulmonary Fibrosis. Diagnostics, 13(5), 935. https://doi.org/10.3390/diagnostics13050935