
Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis
 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. The Aim of the Study
2.2. Regulatory Board Approval
2.3. Study Group
2.4. Principles of HP Diagnosis
- Occupational or environmental exposure to organic antigens was established, and the symptoms of the disease were clearly related to this exposure, and/or positive precipitant immunoglobulins G (IgG) against avian or bacterial antigens were confirmed;
- 2.
- A typical presentation of HP features in HRCT was described (i.e., mosaic lung attenuation, air-trapping, centrilobular nodules, ground glass opacities, upper and middle-lobe predominant fibrosis, peribronchovascular fibrosis, honeycombing) [3];
- 3.
- An increased percentage of lymphocytes exceeding 30% was present in BALF.
2.5. Treatment
2.6. Statistical Analysis
3. Results
3.1. Baseline Characteristics of the Study Group
3.2. Treatment Outcomes
3.3. Treatment Outcome Predictors

{kind=link}
{kind=link}
| Variable | OR | CI | p-Value |
|---|---|---|---|
| Sex | 0.549 | 0.226–1.335 | 0.186 |
| Age at dgn | 0.981 | 0.943–1.020 | 0.338 |
| Time from symptoms onset | 0.997 | 0.989–1.005 | 0.442 |
| Precipitins present | 1.707 | 0.665–4.383 | 0.266 |
| TLC %pred | 1.017 | 0.996–1.038 | 0.113 |
| VC % pred | 1.003 | 0.982–1.025 | 0.787 |
| TL,co % pred | 0.974 | 0.945–1.003 | 0.080 |
| Fever | 11.82 | 1.50–93.26 | 0.019 |
| RV%TLC >120%pred | 3.352 | 1.251–8.983 | 0.016 |
| Lymphocytes in BALF > 53.55% | 8.30 | 2.24–3.79 | 0.001 |
| CT–centrilobular nodules | 3.078 | 1.186–7.983 | 0.021 |
| CT–fibrosis (any) | 0.133 | 0.041–0.425 | <0.001 |
| Eosinophiles in BALF > 2.875% | 0.23 | 0.08–0.67 | 0.0006 |
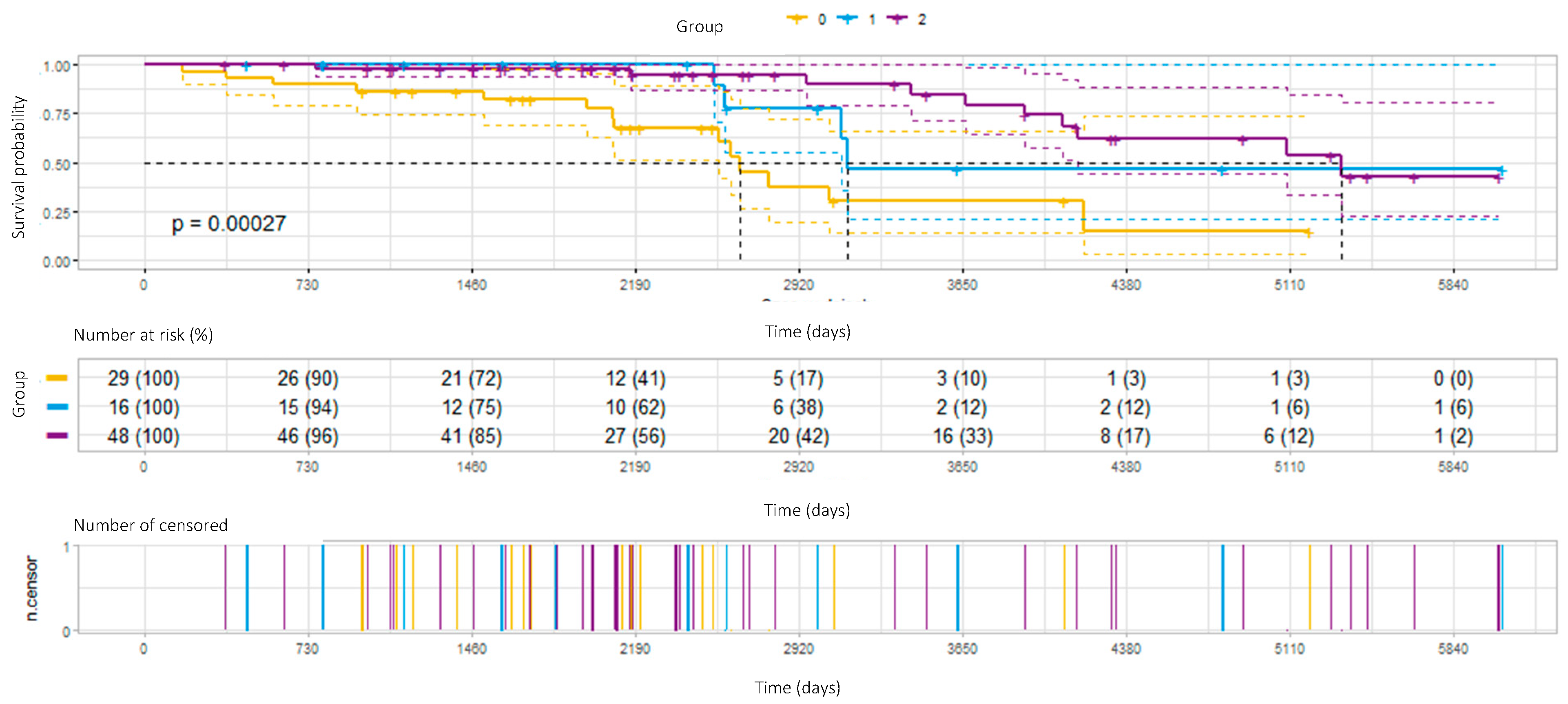
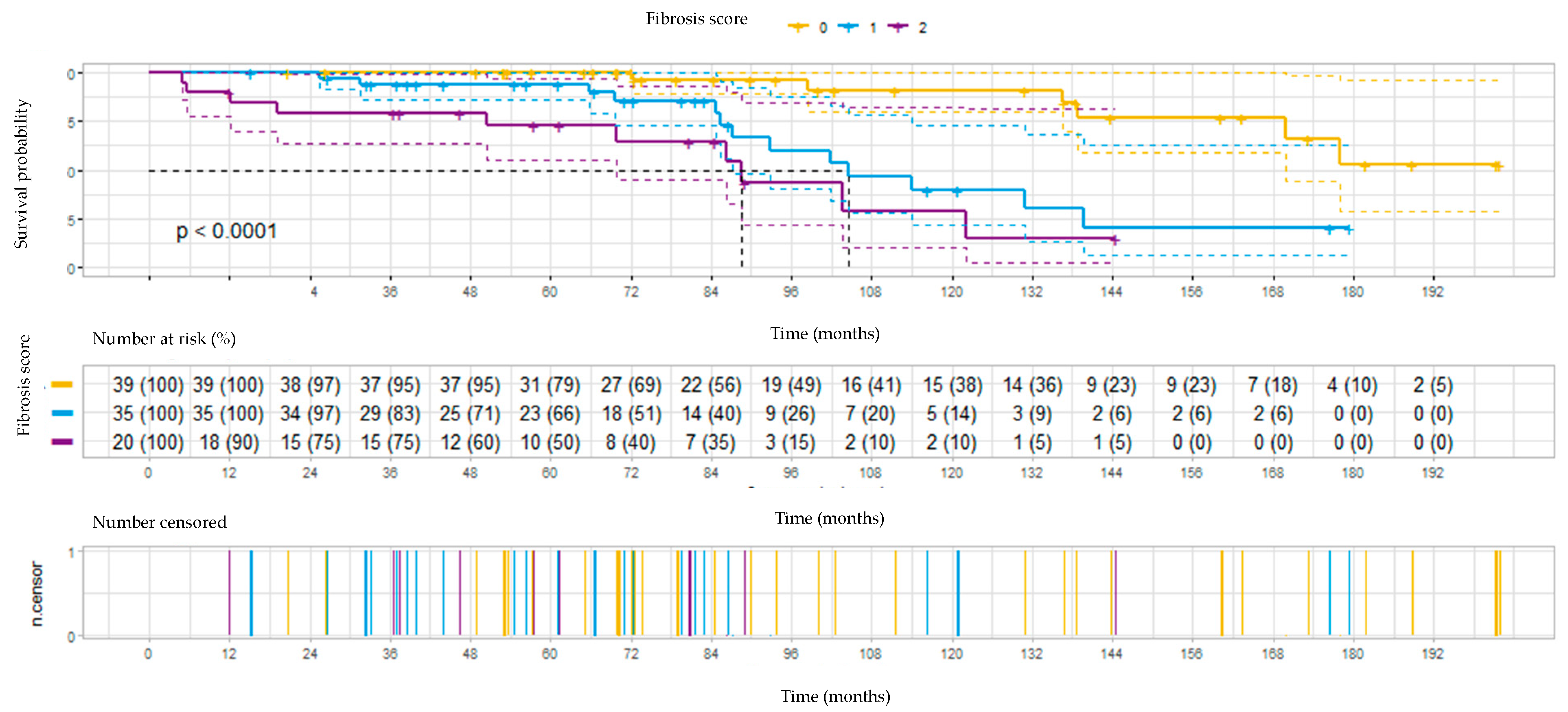
3.4. Survival
4. Discussion
Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef] [PubMed]
- Jędrych, M.E.; Szturmowicz, M.; Bestry, I.; Kuś, J. Hypersensitivity pneumonitis: Diagnostic criteria, treatment, prognosis and prevention. Med. Pr. 2016, 67, 517–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.F.; Sverzellati, N.; Devaraj, A.; Wells, A.U.; Hansell, D.M. Chronic hypersensitivity pneumonitis: High resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur. Radiol. 2012, 22, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Szturmowicz, M.; Barańska, I.; Skoczylas, A.; Jędrych, M.E.; Demkow, U. Correlation of bronchoalveolar lavage lymphocyte count with the extent of lung fibrosis and with plethysmographic lung volumes in patients with newly recognized hypersensitivity pneumonitis. Cent. Eur. J. Immunol. 2020, 45, 276–282. [Google Scholar] [CrossRef]
- De Sadeleer, L.J.; Hermans, F.; De Dycker, E.; Yserbyt, J.; Verschakelen, J.A.; Verbeken, E.K.; Verleden, G.M.; Wuyts, W.A. Effects of Corticosteroid Treatment and Antigen Avoidance in a Large Hypersensitivity Pneumonitis Cohort: A Single-Centre Cohort Study. J. Clin. Med. 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- De Sadeleer, L.J.; Hermans, F.; De Dycker, E.; Yserbyt, J.; Verschakelen, J.A.; Verbeken, E.K.; Verleden, G.M.; Verleden, S.E.; Wuyts, W.A. Impact of BAL lymphocytosis and presence of honeycombing on corticosteroid treatment effect in fibrotic hypersensitivity pneumonitis: A retrospective cohort study. Eur. Respir. J. 2020, 55, 1901983. [Google Scholar] [CrossRef]
- Adegunsoye, A.; Oldham, J.M.; Fernández Pérez, E.R.; Hamblin, M.; Patel, N.; Tener, M.; Bhanot, D.; Robinson, L.; Bullick, S.; Chen, L.; et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 2017, 3, 00016-2017. [Google Scholar] [CrossRef]
- Ejima, M.; Okamoto, T.; Suzuki, T.; Anzai, T.; Takahashi, K.; Miyazaki, Y. Efficacy of treatment with corticosteroids for fibrotic hypersensitivity pneumonitis: A propensity score-matched cohort analysis. BMC Pulm. Med. 2021, 21, 243. [Google Scholar] [CrossRef]
- Tony, F.A.; Soliman, Y.M.A.; Salem, H.A. Effect of Oral Methyl Prednisolone on Different Radiological Patterns of Hypersensitivity Pneumonitis. J. Asthma Allergy 2021, 14, 501–511. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Barańska, I.; Jędrych, M.E.; Bartoszuk, I.; Radwan-Roehrenschef, P.; Roży, A.; Bestry, I.; Chorostowska-Wynimko, J.; Langfort, R.; Kuś, J. Hypersensitivity pneumonitis recognised in a single pulmonary unit, between 2005 and 2015—Comparison with recently proposed diagnostic criteria. Adv. Respir. Med. 2019, 87, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Wanger, J.; Clausen, J.L.; Coates, A.; Pedersen, O.F.; Brusasco, V.; Burgos, F.; Casaburi, R.; Crapo, R.; Enright, P.; van der Grinten, C.P.M.; et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 2005, 26, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.H.; Tammeling, G.J.; Cotes, J.E.; Pedersen, O.F.; Peslin, R.; Yernault, J.C. Lung volumes and forced ventilatory flows. Report Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur. Respir. J. Suppl. 1993, 16, 5–40. [Google Scholar] [CrossRef]
- Graham, B.L.; Brusasco, V.; Burgos, F.; Cooper, B.G.; Jensen, R.; Kendrick, A.; MacIntyre, N.R.; Thompson, B.R.; Wanger, J. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur. Respir. J. 2017, 49, 1600016. [Google Scholar] [CrossRef]
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An Official American Thoracic Society Clinical Practice Guideline: The Clinical Utility of Bronchoalveolar Lavage Cellular Analysis in Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef]
- Morisset, J.; Johannson, K.A.; Vittinghoff, E.; Aravena, C.; Elicker, B.M.; Jones, K.D.; Fell, C.D.; Manganas, H.; Dubé, B.-P.; Wolters, P.J.; et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017, 151, 619–625. [Google Scholar] [CrossRef]
- Fiddler, C.A.; Simler, N.; Thillai, M.; Parfrey, H. Use of mycophenolate mofetil and azathioprine for the treatment of chronic hypersensitivity pneumonitis-A single-centre experience. Clin. Respir. J. 2019, 13, 791–794. [Google Scholar] [CrossRef]
- Raimundo, S.; Pimenta, A.C.; Cruz-Martins, N.; Rodrigues, M.C.; Melo, N.; Mota, P.C.; Sokhatska, O.; Bastos, H.N.; Beltrão, M.; Guimarães, S.; et al. Insights on chronic hypersensitivity pneumonitis’ treatment: Factors associated with a favourable response to azathioprine. Life Sci. 2021, 272, 119274. [Google Scholar] [CrossRef]
- Sterclova, M.; Smetakova, M.; Stehlik, L.; Skibova, J.; Vasakova, M. Bronchoalveolar lavage cell profiles and proteins concentrations can be used to phenotype extrinsic allergic alveolitis patients. Multidiscip. Respir. Med. 2019, 14, 13. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Swigris, J.J.; Forssén, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, M.L.; Gu, T.; Murray, S.; Gross, B.H.; Chughtai, A.; Sayyouh, M.; Kazerooni, E.A.; Myers, J.L.; Lagstein, A.; Konopka, K.E.; et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated with Distinct Survival Time and Pulmonary Function Trajectory. Chest 2019, 155, 699–711. [Google Scholar] [CrossRef] [PubMed]


| Treatment Outcome | Improvement | Stable Disease | Progression | |
|---|---|---|---|---|
| Parameter | ||||
| VC max 1 | Increase by 10% or more | Increase by < 10% | Any decrease | |
| OR | ||||
| TL,co 2 | Increase by 15% or more | Increase by < 15% | Any decrease | |
| AND | ||||
| Chest X-ray changes | Improvement or stable | Improvement or stable | Worsening or stable | |
| Variable | Whole Group N = 93 | Non-Fibrotic HP N = 39 | Fibrotic HP N = 54 | p-Value nf-HP vs. f-HP |
|---|---|---|---|---|
| Age at diagnosis (y), mean (± SD) | 51.7 (±11.69) | 49 (±12.81) | 53.7 (±10.52) | 0.0921 |
| Male, No (%) | 49 (53) | 19 (48.7 *) | 26 (48.15 #) | 0.9567 |
| Ever smoker, No (%) | 39 (41.5) | 12 (30.77 *) | 27 (50 #) | 0.0637 |
| Time from symptoms onset to diagnosis mean (± SD), mo | 34 (± 40.5) | 28.8 (± 62.92) | 39.8 (±45.1) | 0.0004 |
| Fever, No (%) | 20 (21) | 9 (23 *) | 11 (20.3 #) | 0.7539 |
| Antigen exposure, No (%) | ||||
| Poultry | 32 (34) | 17 (43.6 *) | 15 (27.8 #) | 0.1271 |
| Pigeons | 19 (20) | 8 (20.5 *) | 11 (20.4 #) | 0.999 |
| Parrots | 4 (4) | 1 (2.6 *) | 3 (5.6 #) | 0.6368 |
| Hay/feed | 43 (46) | 26 (66.7 *) | 17 (31.5 #) | 0.0015 |
| VC max (L), mean (± SD) | 2.95 (±0.97) | 3.14 (± 0.931) | 2.80 (±0.989) | 0.0729 |
| VC max (% pred.), mean (± SD) | 81.5 (±20.8) | 86.5 (± 19.76) | 78.3 (±20.34) | 0.0622 |
| TLC (L), mean (± SD) | 4.96 (±1.47) | 5.58 (± 1.6) | 4.48 (±1.16) | 0.0006 |
| TLC (% pred.), mean (± SD) | 88.6 (±23.5) | 99.7 (± 24.42) | 78.5 (±16.84) | <0.0001 |
| RV%TLC (% pred.), mean (± SD) | 113.08 (±26.86) | 122.4 (± 32.3) | 106.1(±19.49) | 0.0034 |
| TLco (% pred.), mean (± SD) | 48.3 (±15.7) | 53.2 (± 15.35) | 44.6 (±14.80) | 0.0140 |
| Tiffenau index (%), mean (± SD) | 80.2 (±8.23) | 75.95 (± 13.97) | 80.02 (±7.55) | 0.2691 |
| 6MWD (m), mean (± SD) | 485.7 (±106.3) | 472.9 (± 116.4) | 484.0 (±98.48) | 0.7968 |
| HRCT fibrosis any, No (%) | 54 (58) | 0 | 54 (100 #) | |
| HRCT fibrosis—score 1, No (%) | 35 (38) | 0 | 35 (64.8 #) | |
| HRCT fibrosis—score 2, No (%) | 19 (20) | 0 | 19 (35.2 #) | |
| HRCT ground glass opacities, No (%) | 79 (85) | 35 (89.7 *) | 45 (83.33 #) | 0.5466 |
| HRCT centrilobular nodules, No (%) | 42 (45) | 21(53.85 *) | 22(51.16 #) | 0.2920 |
| BAL performed, No (%) | 78 (83.9) | 33 (84.6 *) | 45 (85.18 #) | 0.999 |
| BALF cells’ count (M), mean (± SD) | 33.59 (±21.92) | 41.47 (±28.1) | 28.13 (±13.67) | 0.0610 |
| BALF lymph (%), mean (± SD) | 47.13 (±19.14) | 59.98 (± 14.26) | 38.48 (±17.10) | <0.0001 |
| BALF neut (%), mean (± SD) | 6.1 (±6.03) | 4.87 (±5.41) | 6.97 (±6.34) | 0.0924 |
| BALF eos (%), mean (± SD) | 2.52 (±3.42) | 1.68 (±2.34) | 3.12 (±3.93) | 0.0431 |
| TBLB performed, No (%) | 29 (31) | 14 (35.9 *) | 15 (26.8 #) | 0.3722 |
| SLB performed, No (%) | 29 (31) | 8 (20.5 *) | 21 (38.9 #) | 0.0719 |
| CS monotherapy, No (%) | 76 (82) | 37 (94.9 *) | 39 (72.2 #) | 0.006 |
| CS + AZA treatment, No (%) | 17 (18) | 2 (5.1 *) | 15 (27.8 #) | 0.006 |
| Characteristics | N (%) | Improvement N (%) | Stable Disease N (%) | Progression N (%) | p |
|---|---|---|---|---|---|
| Whole group | 93 (100) | 49 (53) | 16 (17) | 28 (30) | |
| Non-fibrotic HP | 39 (100) | 30 (77) | 5 (13) | 4 (10) | 0.00069 |
| Fibrotic HP | 54 (100) | 19 (35) | 11 (20) | 24 (45) |
| Variable | OR | 95%CI | p-Value |
|---|---|---|---|
| Fever | 5.973 | 0.8116–125.0 | 0.1278 |
| RV%TLC >120%pred | 1.665 | 0.4462–6.404 | 0.4458 |
| Lymphocytes in BALF >53.55% | 2.048 | 0.3940–11.94 | 0.3958 |
| CT–centrilobular nodules | 1.737 | 0.5104–6.178 | 0.3794 |
| CT–fibrosis (any) | 0.1489 | 0.0261–0.6552 | 0.0178 |
| Eosinophiles in BALF >2.875% | 0.2669 | 0.0643–0.945 | 0.0527 |
| Variable | OR | 95%CI | p-Value |
|---|---|---|---|
| VC max > 65% | 0.330 | 0.155–0.702 | 0.0025 |
| TLC > 72.5% | 0.298 | 0.140–0.633 | 0.002 |
| Improvement after treatment | 0.4511 | 0.293–0.693 | <0.001 |
| Fibrosis in HRCT | 2.945 | 1.813–4.784 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska, K.B.; Barańska, I.; Sobiecka, M.; Radwan-Rohrenschef, P.; Dybowska, M.; Franczuk, M.; Roży, A.; Skoczylas, A.; Bestry, I.; Kuś, J.; et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis. Diagnostics 2022, 12, 2767. https://doi.org/10.3390/diagnostics12112767
Lewandowska KB, Barańska I, Sobiecka M, Radwan-Rohrenschef P, Dybowska M, Franczuk M, Roży A, Skoczylas A, Bestry I, Kuś J, et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis. Diagnostics. 2022; 12(11):2767. https://doi.org/10.3390/diagnostics12112767
Chicago/Turabian StyleLewandowska, Katarzyna B., Inga Barańska, Małgorzata Sobiecka, Piotr Radwan-Rohrenschef, Małgorzata Dybowska, Monika Franczuk, Adriana Roży, Agnieszka Skoczylas, Iwona Bestry, Jan Kuś, and et al. 2022. "Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis" Diagnostics 12, no. 11: 2767. https://doi.org/10.3390/diagnostics12112767
APA StyleLewandowska, K. B., Barańska, I., Sobiecka, M., Radwan-Rohrenschef, P., Dybowska, M., Franczuk, M., Roży, A., Skoczylas, A., Bestry, I., Kuś, J., Tomkowski, W. Z., & Szturmowicz, M. (2022). Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis—Retrospective Cohort Analysis. Diagnostics, 12(11), 2767. https://doi.org/10.3390/diagnostics12112767