
Genotypic Frequencies of Mutations Associated with Alpha-1 Antitrypsin Deficiency in Unrelated Bone Marrow Donors from the Murcia Region Donor Registry in the Southeast of Spain

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
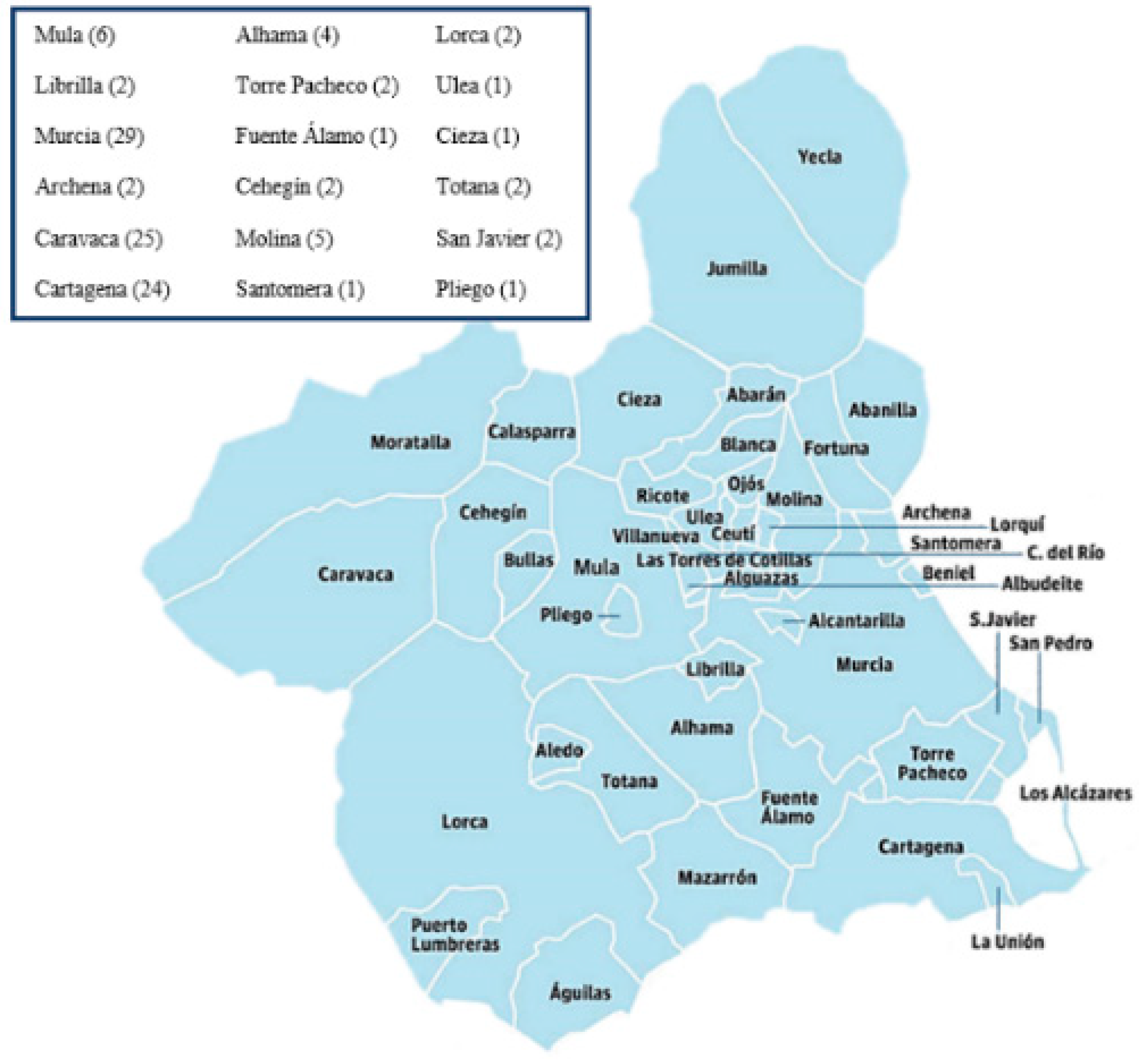
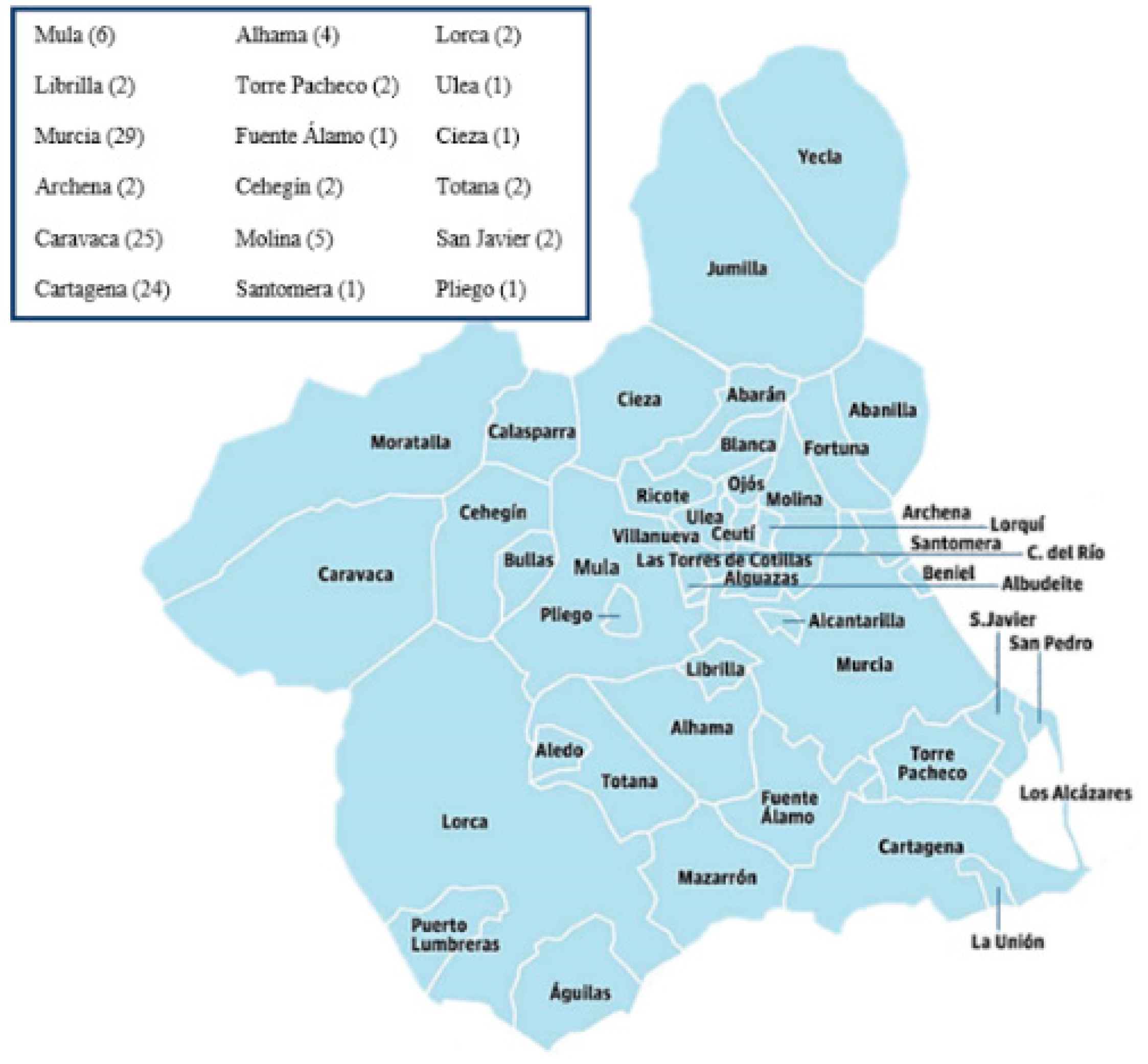
2.1. Demographic Information, Clinical Variables, and Study Methodology
2.2. Genomic Extraction
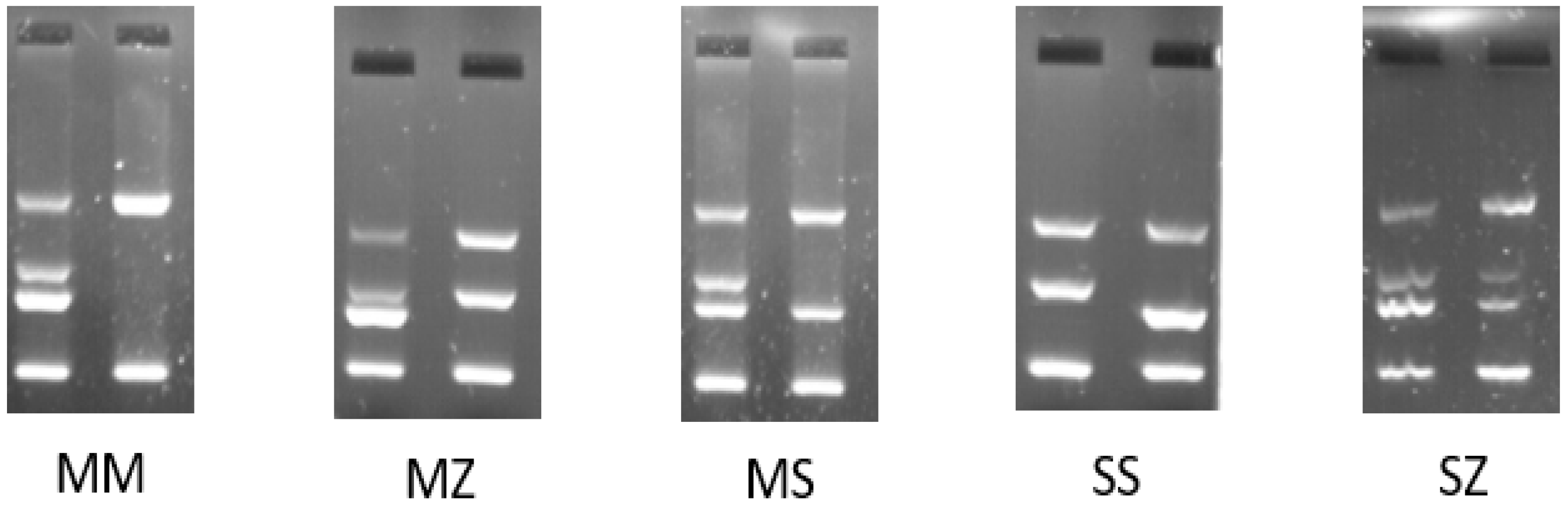
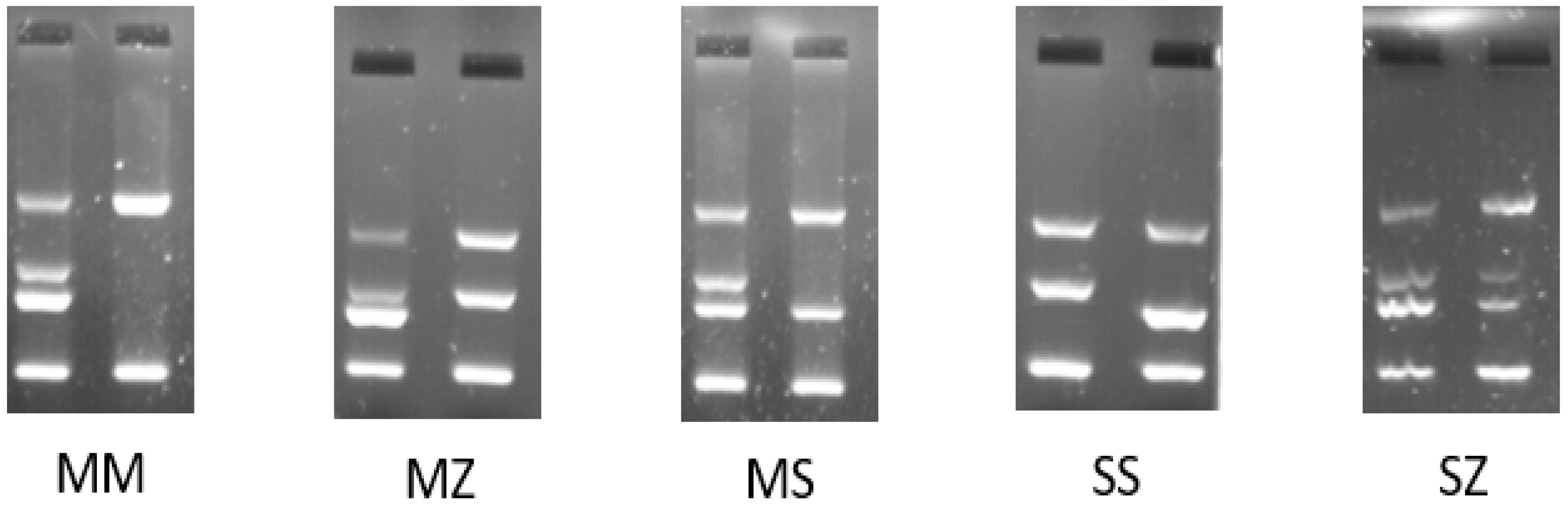
2.3. PCR Amplification-Refractory Mutation System (ARMS-PCR)
2.4. Statistical Analysis
3. Results
3.1. AAT1 Genotype Analysis
3.2. Analysis of Alleles of the Pi*Z and Pi*S Variants in the SERPINE1 Gene
3.3. Analysis of Genotype Frequencies of Pi*Z (E342K) and Pi*S (E264V) AAT1 Mutations
3.4. Analysis of Haplotype Frequencies of E342K and E264V in the SERPINE1 Gene
3.5. Estimation of the AAT1 Pi Genotype in the Murcian Population
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hubbard, R.C.; Ogushi, F.; Fells, G.A.; Cantin, A.M.; Jallat, S.; Courtney, M.; Crystal, R.G. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of alpha 1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J. Clin. Investig. 1987, 80, 1289–1295. [Google Scholar] [CrossRef]
- Bucurenci, N.; Blake, D.R.; Chidwick, K.; Winyard, P.G. Inhibition of neutrophil superoxide production by human plasma alpha 1-antitrypsin. FEBS Lett. 1992, 300, 21–24. [Google Scholar] [CrossRef]
- Vidal, R.; Blanco, I.; Casas, F.; Jardí, R.; Miratvilles, M.; Barros-Tizón, J.C.; Bustamante, A.; Escudero, C.; España, P.P.; Martínez, M.; et al. Diagnóstico y tratamiento del déficit de alfa-1-antitripsina. Arch. Bronconeumol. 2006, 42, 645–659. [Google Scholar] [CrossRef]
- Forney, J.R.; Yang, S.; Healey, M.C. Antagonistic effect of human alpha-1-antitrypsin on excystation of Cryptosporidium parvum oocysts. J. Parasitol. 1997, 83, 771–774. [Google Scholar] [CrossRef]
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.U.; Dafforn, T.R.; Stockley, R.A. α-1-antitrypsin variants and the proteinase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L179–L190. [Google Scholar] [CrossRef]
- Duranton, J.; Bieth, J.G. Inhibition of proteinase 3 by [alpha]1-antitrypsin in vVitro predicts very fast inhibition in vivo. Am. J. Respir. Cell Mol. Biol. 2003, 29, 57–61. [Google Scholar] [CrossRef]
- Duranton, J.; Adam, C.; Bieth, J.G. Kinetic mechanism of the inhibition of cathepsin G by alpha 1-antichymotrypsin and alpha 1-proteinase inhibitor. Biochemistry 1998, 37, 11239–11245. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.L.; Bailo, M.; Reihill, J.A.; Crilly, A.; Lockhart, J.C.; Litherland, G.J.; Lundy, F.T.; McGarvey, L.P.; Hollywood, M.A.; Martin, S.L. Trypsin-Like Proteases and Their Role in Muco-Obstructive Lung Diseases. Int. J. Mol. Sci. 2021, 22, 5817. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.J.; Tarran, R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Int. J. Mol. Sci. 2018, 22, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Lomas, D.A. Alpha 1 -Antitrypsin Deficiency—A Model for Conformational Diseases. N. Engl. J. Med. 2002, 346, 45–53. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-Antitrypsin deficiency. Nat. Rev. Dis. Prim. 2016, 28, 16051. [Google Scholar] [CrossRef]
- Fagerhol, M.K.; Laurell, C.B. The polymorphism of “prealbumins” and α1-antitrypsin in human sera. Clin. Chim. Acta 1967, 16, 199–203. [Google Scholar] [CrossRef]
- Stolz, D.; Mkorombindo, T.; Schumann, D.M.; Agusti, A.; Ash, S.Y.; Bafadhel, M.; Bai, C.; Chalmers, J.D.; Criner, G.J.; Dharmage, S.C.; et al. Towards the elimination of chronic obstructive pulmonary disease: A Lancet Commission. Lancet 2022, 400, 921–972. [Google Scholar] [CrossRef]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A Genome-Wide Association Study in Chronic Obstructive Pulmonary Disease (COPD): Identification of Two Major Susceptibility Loci. PLOS Genet. 2009, 5, e1000421. [Google Scholar] [CrossRef]
- DeMeo, D.L.; Silverman, E.K. α1-Antitrypsin deficiency · 2: Genetic aspects of α1-antitrypsin deficiency: Phenotypes and genetic modifiers of emphysema risk. Thorax 2004, 59, 259–264. [Google Scholar] [CrossRef]
- Ioachimescu, O.C.; Stoller, J.K. A Review of Alpha-1 Antitrypsin Deficiency. COPD J. Chronic Obstr. Pulm. Dis. 2009, 2, 263–275. [Google Scholar] [CrossRef]
- de Serres, F.J. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ. Health Perspect. 2003, 111, 1851–1854. [Google Scholar] [CrossRef]
- DeMeo, D.L.; Sandhaus, R.A.; Barker, A.F.; Brantly, M.L.; Eden, E.; McElvaney, N.G.; Rennard, S.; Burchard, E.; Stocks, J.M.; Stoller, J.K.; et al. Determinants of airflow obstruction in severe alpha-1-antitrypsin deficiency. Thorax 2007, 62, 806–813. [Google Scholar] [CrossRef]
- Chua, F.; Laurent, G.J. Neutrophil elastase: Mediator of extracellular matrix destruction and accumulation. Proc. Am. Thorac. Soc. 2006, 3, 424–427. [Google Scholar] [CrossRef]
- Janus, E.D.; Phillips, N.T.; Carrell, R.W. Smoking, Lung Function, and A1-Antitrypsin Deficiency. Lancet 1985, 325, 152–154. [Google Scholar] [CrossRef]
- Miravitlles, M.; Dirksen, A.; Ferrarotti, I.; Koblizek, V.; Lange, P.; Mahadeva, R.; McElvaney, N.G.; Parr, D.; Piitulainen, E.; Roche, N.; et al. European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. Eur. Respir. J. 2017, 50, 1700610. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society/European Respiratory Society statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [CrossRef] [PubMed]
- Fagerhol, M.K.; Hauge, H.E. The Pi Phenotype MP. Vox Sang. 1968, 15, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Conde, G.; Lara, B.; Miravitlles, M. Augmentation therapy for emphysema due to alpha-1-antitrypsin deficiency. Ther. Adv. Respir. Dis. Rev. Ther. Adv. Respir. Dis. 2008, 2, 13–21. [Google Scholar] [CrossRef] [PubMed]
- WMA Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects—WMA—The World Medical Association. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 9 March 2023).
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-antitrypsin deficiency: Genetic variations, clinical manifestations and therapeutic interventions. Mutat. Res. Mutat. Res. 2017, 773, 14–25. [Google Scholar] [CrossRef]
- Curiel, D.T.; Chytil, A.; Courtney, M.; Crystal, R.G. Serum α1-antitrypsin deficiency associated with the common S-type (Glu264 → Val) mutation results from intracellular degradation of α1-antitrypsin prior to secretion. J. Biol. Chem. 1989, 264, 10477–10486. [Google Scholar] [CrossRef]
- Ogushi, F.; Straus, S.D.; Crystal, R.G. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. Find the latest version: Z-Type al-Antitrypsin Is Less Competent Than Mi-Type al -Antitrypsin as an Inhibitor of Neutrophil Elastase. J. Immunol. 1987, 80, 1366–1374. [Google Scholar]
- Boix, F.; Legaz, I.; Minhas, A.; Alfaro, R.; Jiménez–Coll, V.; Mrowiec, A.; Martínez–Banaclocha, H.; Galián, J.A.; Botella, C.; Moya–Quiles, M.R.; et al. Identification of peripheral CD154 + T cells and HLA-DRB1 as biomarkers of acute cellular rejection in adult liver transplant recipients. Clin. Exp. Immunol. 2021, 203, 315–328. [Google Scholar] [CrossRef]
- Cox, D.W. Genetic variation of alpha 1-antitrypsin. Am. J. Hum. Genet. 1978, 30, 660. [Google Scholar]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The discovery of α1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- De La Roza, C.; Lara, B.; Vilà, S.; Miravitlles, M. Déficit de alfa-1-antitripsina. Situación en España y desarrollo de un programa de detección de casos. Arch. Bronconeumol. 2006, 42, 290–298. [Google Scholar] [CrossRef] [PubMed]
- EBSCOhost | 121383424 | Understanding Alpha-1 Antitrypsin Deficiency: A Review with an Allergist’s Outlook. Available online: https://web.s.ebscohost.com/abstract?direct=true&profile=ehost&scope=site&authtype=crawler&jrnl=10885412&asa=Y&AN=121383424&h=nGemLTG%2BNARDdhHm%2B4hsgWh9Jk5R7B9k3JPtc9vzlIXqcIax3nbRLPIzBBLeepH5jQzLYVwUS8Y%2BchAqZrbq1A%3D%3D&crl=c&resultNs=AdminWebAuth&resultLocal=ErrCrlNotAuth&crlhashurl=login.aspx%3Fdirect%3Dtrue%26profile%3Dehost%26scope%3Dsite%26authtype%3Dcrawler%26jrnl%3D10885412%26asa%3DY%26AN%3D121383424 (accessed on 9 March 2023).
- de Serres, F.; Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Herr, C.; Ferrarotti, I.; Jardi, R.; Rodriguez-Frias, F.; Luisetti, M.; Bals, R. Laboratory testing of individuals with severe α1-antitrypsindeficiency in three European centres. Eur. Respir. J. 2010, 35, 960–968. [Google Scholar] [CrossRef] [PubMed]
- de Serres, F.J.; Blanco, I.; Fernández-Bustillo, E. Estimated numbers and prevalence of PI*S and PI*Z deficiency alleles of α1-antitrypsin deficiency in Asia. Eur. Respir. J. 2006, 28, 1091–1099. [Google Scholar] [CrossRef]
- Muro, M.; Moya-Quiles, M.R.; Botella, C.; García, L.; Minguela, A.; Álvarez-López, M.R. Genetic relationship between Murcia Region (SE Spain) and other populations in the Iberian Peninsula and Mediterranean area with respect to HFE gene mutations distribution. Ann. Hematol. 2007, 86, 455–457. [Google Scholar] [CrossRef]
- Muro, M.; Marín, L.; Torío, A.; Moya-Quiles, M.R.; Minguela, A.; Rosique-Roman, J.; Sanchis, M.J.; Garcia-Calatayud, M.C.; García-Alonso, A.M.; Álvarez-López, M.R. HLA polymorphism in the murcia population (Spain): In the cradle of the archaeologic Iberians. Hum. Immunol. 2001, 62, 910–921. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Variants | Exon | Mutation | Variation | Reference |
|---|---|---|---|---|
| S | IV | GAA → GTG | Glu264 → Val264 (E264V) | [27] |
| Z | V | GAG → AAG | Glu342 → Lys342 (E342K) | [28] |
| SERPINE1 Gene | Total N = 112; 2n = 224 N (%) | 95% CI (%) |
|---|---|---|
| Allele frequencies/variation | ||
| GAG/E342 (wild-type) | 217 (96.8%) | 95.9–98.7 |
| AAG/K342 (mutant) | 7 (3.12%) | 1.41–3.99 |
| GAA/E264 (wild-type) | 199 (88.8%) | 75.6–89.6 |
| GTG/V264 (mutant) | 25 (11.1%) | 8.81–12.41 |
| Genotype frequencies | ||
| EE342 | 105 (93.75%) | 92.8–95.2 |
| EK342 | 7 (6.25%) | 4.0–8.8 |
| KK342 | 0 (0.0%) | 0 |
| EE264 | 88 (78.57%) | 70.3–81.8 |
| EV264 | 23 (20.53%) | 18.7–23.6 |
| VV264 | 1 (0.89%) | 0.4–2.79 |
| Haplotype Frequencies of SERPINE1 | Total N = 112; 2n = 224 (%) N (%) | 95% CI (%) |
|---|---|---|
| EE342 EE264 | 83 (74.10%) | 71.7–81.1 |
| EK342 EE264 | 5 (4.46%) | 4.0–5.9 |
| KK342 EE264 | 0 (0.0%) | 0 |
| EE342 EV264 | 21 (18.75%) | 17.8–22.1 |
| EE342 VV264 | 1 (0.89%) | 0.81–1.1 |
| EK342 EV264 | 2 (1.78%) | 1.7–2.1 |
| EK342 VV264 | 0 (0.0%) | 0 |
| KK342 EV264 | 0 (0.0%) | 0 |
| KK342 VV264 | 0 (0.0%) | 0 |
| Pi* Genotype | ||
| Pi*MM | 83 (74.10%) | 71.7–84.1 |
| Pi*MZ | 5 (4.46%) | 3.9–5.99 |
| Pi*ZZ | 0 (0.0%) | 0 |
| Pi*MS | 21 (18.75%) | 17.8–22.1 |
| Pi*SS | 1 (0.89%) | 0.71–2.11 |
| Pi*SZ | 2 (1.78%) | 1.07–2.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuenca, I.; Botella, C.; Moya-Quiles, M.R.; Jimenez-Coll, V.; Galian, J.A.; Martinez-Banaclocha, H.; Muro-Pérez, M.; Minguela, A.; Legaz, I.; Muro, M. Genotypic Frequencies of Mutations Associated with Alpha-1 Antitrypsin Deficiency in Unrelated Bone Marrow Donors from the Murcia Region Donor Registry in the Southeast of Spain. Diagnostics 2023, 13, 2845. https://doi.org/10.3390/diagnostics13172845
Cuenca I, Botella C, Moya-Quiles MR, Jimenez-Coll V, Galian JA, Martinez-Banaclocha H, Muro-Pérez M, Minguela A, Legaz I, Muro M. Genotypic Frequencies of Mutations Associated with Alpha-1 Antitrypsin Deficiency in Unrelated Bone Marrow Donors from the Murcia Region Donor Registry in the Southeast of Spain. Diagnostics. 2023; 13(17):2845. https://doi.org/10.3390/diagnostics13172845
Chicago/Turabian StyleCuenca, Irene, Carmen Botella, María Rosa Moya-Quiles, Víctor Jimenez-Coll, José Antonio Galian, Helios Martinez-Banaclocha, Manuel Muro-Pérez, Alfredo Minguela, Isabel Legaz, and Manuel Muro. 2023. "Genotypic Frequencies of Mutations Associated with Alpha-1 Antitrypsin Deficiency in Unrelated Bone Marrow Donors from the Murcia Region Donor Registry in the Southeast of Spain" Diagnostics 13, no. 17: 2845. https://doi.org/10.3390/diagnostics13172845
APA StyleCuenca, I., Botella, C., Moya-Quiles, M. R., Jimenez-Coll, V., Galian, J. A., Martinez-Banaclocha, H., Muro-Pérez, M., Minguela, A., Legaz, I., & Muro, M. (2023). Genotypic Frequencies of Mutations Associated with Alpha-1 Antitrypsin Deficiency in Unrelated Bone Marrow Donors from the Murcia Region Donor Registry in the Southeast of Spain. Diagnostics, 13(17), 2845. https://doi.org/10.3390/diagnostics13172845