
FDG-PET versus Amyloid-PET Imaging for Diagnosis and Response Evaluation in Alzheimer’s Disease: Benefits and Pitfalls

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Clinical Efficacy
3. PET Imaging in General
4. The Diagnosis of AD or Alzheimer’s Syndrome
4.1. Before the Advent of Amyloid-PET Imaging
4.2. After the Advent of Amyloid-PET Imaging
5. The Role of PET Imaging
5.1. Amyloid-PET Imaging
5.2. FDG-PET Imaging
6. Recommendations
- The diagnosis of AD/AS should be based upon co-occurrence of the following findings: (a) impaired cognitive function on the MMSE and/or CDR-SB scale, assessed by a trained neuropsychologist, (b) impaired temporo-parietal glucose metabolism assessed by FDG-PET according to standardized imaging and analysis procedures, (c) absence of other well-defined disorders, including tumor, metastases, trauma, and stroke, (d) absence of clinical disease phenotypes closely associated with frontotemporal lobar degeneration and young-onset dementia, excluded by standardized criteria, but not necessarily absence of vascular dementia.
- Positive therapy efficacy equals: (a) favorable change (exceeding an a priori predefined minimum limit) in cognitive ability as measured on a recognized cognitive scale, (b) increased global or specified regional cerebral metabolism assessed by repeat FDG-PET brain imaging, and (c) less decrease in global brain and hippocampal volumes and less increase in ventricular volume assessed by volume MRI.
- Registration and neuropathologic examination of all deaths occurring during and two years after termination of clinical trials should be carried out.
- The limited CMS reimbursement for amyloid-PET-scans in dementia patients should not be changed until results of phase 4 confirmatory immunotherapy trials are available.
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Shumeyko, S.A.; Vikhlyantsev, I.M.; Bobylev, A.G. Amyloids: The History of Toxicity and Functionality. Biology 2021, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration; Cavazzoni, P. FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 12 May 2023).
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 12 May 2023).
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Eisai Global. Lecanemab Confirmatory Phase 3 Clarity AD Study Met Primary Endpoint, Showing Highly Statistically Significant Reduction of Clinical Decline in Large Global Clinical Study of 1,795 Participants with Early Alzheimer’s Disease. Available online: https://www.eisai.com/news/2022/news202271.html (accessed on 17 May 2023).
- Cision PR Newswire. Lilly’s Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease. Available online: prnewswire.com (accessed on 17 May 2023).
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderbman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Andrews, J.S.; Desai, U.; Kirson, N.Y.; Zichlin, M.L.; Ball, D.E.; Matthews, B. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer’s disease clinical trials. Alzheimers Dement. 2019, 5, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.M.; Rentz, D.M.; Case, M.; Lauzon, S.; Sims, J.R. Integrated Alzheimer’s Disease Rating Scale: Clinically meaningful change estimates. Alzheimers Dement. 2022, 8, e12312. [Google Scholar] [CrossRef]
- Kastelik-Hryniewiecka, A.; Jewula, P.; Bakalorz, K.; Kramer-Marek, G.; Kuźnik, N. Targeted PET/MRI Imaging Super Probes: A Critical Review of Opportunities and Challenges. Int. J. Nanomed. 2022, 16, 8465–8483. [Google Scholar] [CrossRef] [PubMed]
- Høilund-Carlsen, P.F.; Edenbrandt, L.; Alavi, A. Global disease score (GDS) is the name of the game! Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1768–1772. [Google Scholar] [CrossRef] [PubMed]
- Høilund-Carlsen, P.F.; Piri, R.; Constantinescu, C.; Iversen, K.K.; Werner, T.J.; Sturek, M.; Alavi, A.; Gerke, O. Atherosclerosis Imaging with 18F-Sodium Fluoride PET. Diagnostics 2020, 10, 852. [Google Scholar] [CrossRef]
- Basu, S.; Hess, S.; Nielsen Braad, P.E.; Olsen, B.B.; Inglev, S.; Høilund-Carlsen, P.F. The Basic Principles of FDG-PET/CT Imaging. PET Clin. 2014, 9, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Høilund-Carlsen, P.F.; Piri, R.; Gerke, O.; Edenbrandt, L.; Alavi, A. Assessment of Total-Body Atherosclerosis by PET/Computed Tomography. PET Clin. 2021, 16, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Kepe, V.; Moghbel, M.C.; Långström, B.; Zaidi, H.; Vinters, H.V.; Huang, S.C.; Satyamurthy, N.; Doudet, D.; Mishani, E.; Cohen, R.M.; et al. Amyloid-β positron emission tomography imaging probes: A critical review. J. Alzheimers Dis. 2013, 36, 613–631. [Google Scholar] [CrossRef]
- Høilund-Carlsen, P.F.; Revheim, M.E.; Alavi, A.; Satyamurthy, N.; Barrio, J.R. Amyloid PET: A Questionable Single Primary Surrogate Efficacy Measure on Alzheimer Immunotherapy Trials. J. Alzheimers Dis. 2022, 90, 1395–1399. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Jagust, W.; Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Haeberlein, S.B.; Holtzman, D.M.; Jessen, F.; Karlawish, J.; Liu, E.; Molinuevo, J.L.; et al. “Alzheimer’s disease” is neither “Alzheimer’s clinical syndrome” nor “dementia”. Alzheimers Dement. 2019, 15, 153–157. [Google Scholar] [CrossRef]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Fur Psychiatr. Psych. Gerichtl. Medizin. 1907, 64, 146–148. [Google Scholar]
- Alzheimer, A. Die Colloidentartung des Gehirns. Arch. Für Psychiatr. Nervenkrankh. 1897, 30, 18–53. [Google Scholar] [CrossRef]
- Tagarelli, A.; Piro, A.; Tagarelli, G.; Lagonia, P.; Quattrone, A. Alois Alzheimer: A Hundred Years after the Discovery of the Eponymous Disorder. Int. J. Biomed Sci. 2006, 2, 196–204. [Google Scholar] [PubMed]
- Lafora, G.R. Über das Vorkommen amyloider Körperchen im Innern der Ganglienzellen. Virchows Arch. Path Anat. 1911, 205, 295–303. [Google Scholar] [CrossRef]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef]
- Knopman, D.S.; Petersen, R.C.; Jack, C.R., Jr. A brief history of “Alzheimer disease”: Multiple meanings separated by a common name. Neurology 2019, 92, 1053–1059. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Payoux, P.; Barré, L.; Desgranges, B.; Voisin, T.; Tauber, C.; La Joie, R.; Tafani, M.; Hommet, C.; Chételat, G.; et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Sabbagh, M.N.; Seibyl, J.; Barthel, H.; Akatsu, H.; Ouchi, Y.; Senda, K.; Murayama, S.; Ishii, K.; Takao, M.; et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. Alzheimers Dement. 2015, 11, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.M.; Waddell, B.; Khan, Z.; Pal, S. A systematic review and meta-analysis of (18)F-labeled amyloid imaging in Alzheimer’s disease. Alzheimers Dement. 2015, 1, 5–13. [Google Scholar] [CrossRef]
- Morris, E.; Chalkidou, A.; Hammers, A.; Peacock, J.; Summers, J.; Keevil, S. Diagnostic accuracy of (18)F amyloid PET tracers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 374–385. [Google Scholar] [CrossRef]
- Salloway, S.; Gamez, J.E.; Singh, U.; Sadowsky, C.H.; Villena, T.; Sabbagh, M.N.; Beach, T.G.; Duara, R.; Fleisher, A.S.; Frey, K.A.; et al. Performance of [18F]flutemetamol amyloid imaging against the neuritic plaque component of CERAD and the current (2012) NIA-AA recommendations for the neuropathologic diagnosis of Alzheimer’s disease. Alzheimers Dement. 2017, 9, 25–34. [Google Scholar] [CrossRef]
- Laforce, R., Jr.; Soucy, J.P.; Sellami, L.; Dallaire-Théroux, C.; Brunet, F.; Bergeron, D.; Miller, B.L.; Ossenkoppele, R. Molecular imaging in dementia: Past, present, and future. Alzheimers Dement. 2018, 14, 1522–1552. [Google Scholar] [CrossRef]
- Suppiah, S.; Didier, M.A.; Vinjamuri, S. The Who, When, Why, and How of PET Amyloid Imaging in Management of Alzheimer’s Disease-Review of Literature and Interesting Images. Diagnostics 2019, 9, 65. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Gatsonis, C.; Apgar, C.; Chaudhary, K.; Gareen, I.; Hanna, L.; Hendrix, J.; Hillner, B.E.; Olson, C.; Lesman-Segev, O.H.; et al. Association of Amyloid Positron Emission Tomography with Subsequent Change in Clinical Management Among Medicare Beneficiaries with Mild Cognitive Impairment or Dementia. JAMA 2019, 321, 1286–1294. [Google Scholar] [CrossRef]
- Vera, J.H.; Eftychiou, N.; Schuerer, M.; Rullmann, M.; Barthel, H.; Sabri, O.; Gisslen, M.; Zetterberg, H.; Blennow, K.; O’Brien, C.; et al. Clinical Utility of β-Amyloid PET Imaging in People Living with HIV With Cognitive Symptoms. J. Acquir. Immune Defic. Syndr. 2021, 87, 826–833. [Google Scholar] [CrossRef]
- Hu, Y.; Kirmess, K.M.; Meyer, M.R.; Rabinovici, G.D.; Gatsonis, C.; Siegel, B.A.; Whitmer, R.A.; Apgar, C.; Hanna, L.; Kanekiyo, M.; et al. Assessment of a Plasma Amyloid Probability Score to Estimate Amyloid Positron Emission Tomography Findings Among Adults with Cognitive Impairment. JAMA Netw. Open 2022, 5, e228392. [Google Scholar] [CrossRef] [PubMed]
- Chapleau, M.; Iaccarino, L.; Soleimani-Meigooni, D.; Rabinovici, G.D. The Role of Amyloid PET in Imaging Neurodegenerative Disorders: A Review. J. Nucl. Med. 2022, 63 (Suppl. 1), 13S–19S. [Google Scholar] [CrossRef]
- Altomare, D.; Barkhof, F.; Caprioglio, C.; Collij, L.E.; Scheltens, P.; Lopes Alves, I.; Bouwman, F.; Berkhof, J.; van Maurik, I.S.; Garibotto, V.; et al. Clinical Effect of Early vs Late Amyloid Positron Emission Tomography in Memory Clinic Patients: The AMYPAD-DPMS Randomized Clinical Trial. JAMA Neurol. 2023, 8, e230997. [Google Scholar] [CrossRef] [PubMed]
- Wassef, H.R.; Colletti, P.M. Commentary: Aducanumab-related ARIA: Paean or lament? Clin. Nucl. Med. 2022, 47, 707–709. [Google Scholar] [CrossRef]
- Høilund-Carlsen, P.F.; Alavi, A.; Revheim, M.E. Re: Aducanumab-Related ARIA: Paean or Lament? Clin. Nucl. Med. 2023, 48, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Høilund-Carlsen, P.F.; Barrio, J.R.; Gjedde, A.; Werner, T.J.; Alavi, A. Circular Inference in Dementia Diagnostics. J. Alzheimers Dis. 2018, 63, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Therneau, T.M.; Weigand, S.D.; Wiste, H.J.; Knopman, D.S.; Vemuri, P.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol. 2019, 76, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Blount, L.G. Better Access to PET Scans Can Help Reduce Racial Inequities in Alzheimer’s Disease-STAT. Available online: statnews.com (accessed on 1 June 2023).
- Høilund-Carlsen, P.F.; Alavi, A.; Barrio, J.R. Better Access to Amyloid-PET Scans Won’t Reduce Racial Inequities in Alzheimer’s Disease. Available online: https://www.statnews.com/2022/11/14/better-access-amyloid-pet-scans-alzheimers-inequities (accessed on 1 June 2023).
- Surmak, A.J.; Wong, K.P.; Cole, G.B.; Hirata, K.; Aabedi, A.A.; Mirfendereski, O.; Mirfendereski, P.; Yu, A.S.; Huang, S.C.; Ringman, J.M.; et al. Probing Estrogen Sulfotransferase-Mediated Inflammation with [11C]-PiB in the Living Human Brain. J. Alzheimers Dis. 2020, 73, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef]
- Zeydan, B.; Schwarz, C.G.; Lowe, V.J.; Reid, R.I.; Przybelski, S.A.; Lesnick, T.G.; Kremers, W.K.; Senjem, M.L.; Gunter, J.L.; Min, H.K.; et al. Investigation of white matter PiB uptake as a marker of white matter integrity. Ann. Clin. Transl. Neurol. 2019, 6, 678–688. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Arenaza-Urquijo, E.M.; Knopman, D.S.; Schwarz, C.G.; Brown, R.D.; Rabinstein, A.A.; Gunter, J.L.; Senjem, M.L.; Przybelski, S.A.; Lesnick, T.; et al. White matter hyperintensities: Relationship to amyloid and tau burden. Brain 2019, 142, 2483–2491. [Google Scholar] [CrossRef]
- Weaver, N.A.; Doeven, T.; Barkhof, F.; Biesbroek, J.M.; Groeneveld, O.N.; Kuijf, H.J.; Prins, N.D.; Scheltens, P.; Teunissen, C.E.; van der Flier, W.M.; et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol. Aging 2019, 84, 225–234. [Google Scholar] [CrossRef]
- Pietroboni, A.M.; Colombi, A.; Carandini, T.; Sacchi, L.; Fenoglio, C.; Marotta, G.; Arighi, A.; De Riz, M.A.; Fumagalli, G.G.; Castellani, M.; et al. Amyloid PET imaging and dementias: Potential applications in detecting and quantifying early white matter damage. Alzheimers Res. Ther. 2022, 14, 33. [Google Scholar] [CrossRef]
- Luo, J.; Ma, Y.; Agboola, F.J.; Grant, E.; Morris, J.C.; McDade, E.; Fagan, A.M.; Benzinger, T.L.S.; Hassenstab, J.; Bateman, R.J.; et al. Longitudinal Relationships of White Matter Hyperintensities and Alzheimer Disease Biomarkers Across the Adult Lifespan. Neurology 2023. [Google Scholar] [CrossRef] [PubMed]
- Ottoy, J.; Ozzoude, M.; Zukotynski, K.; Kang, M.S.; Adamo, S.; Scott, C.; Ramirez, J.; Swardfager, W.; Lam, B.; Bhan, A.; et al. Amyloid-PET of the white matter: Relationship to free water, fiber integrity, and cognition in patients with dementia and small vessel disease. J. Cereb. Blood Flow Metab. 2023, 43, 921–936. [Google Scholar] [CrossRef]
- Jacobs, H.I.L.; Hopkins, D.A.; Mayrhofer, H.C.; Bruner, E.; van Leeuwen, F.W.; Raaijmakers, W.; Schmahmann, J.D. The cerebellum in Alzheimer’s disease: Evaluating its role in cognitive decline. Brain 2018, 141, 37–47. [Google Scholar] [CrossRef]
- Segtnan, E.A.; Majdi, A.; Constantinescu, C.; Grupe, P.; Gerke, O.; Dali, H.F.; Strøm, O.E.; Holm, J.; Alavi, A.; Sadigh-Eteghad, S.; et al. Diagnostic manifestations of total hemispheric glucose metabolism ratio in neuronal network diaschisis: Diagnostic implications in Alzheimer’s disease and mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1164–1174. [Google Scholar] [CrossRef]
- Provost, K.; La Joie, R.; Strom, A.; Iaccarino, L.; Edwards, L.; Mellinger, T.J.; Pham, J.; Baker, S.L.; Miller, B.L.; Jagust, W.J. Crossed cerebellar diaschisis on 18F-FDG PET: Frequency across neurodegenerative syndromes and association with 11C-PIB and 18F-Flortaucipir. J. Cereb. Blood Flow Metab. 2021, 41, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Dumba, M.; Khan, S.; Patel, N.; Perry, L.; Malhotra, P.; Perry, R.; Nijran, K.; Barwick, T.; Wallitt, K.; Win, Z. Clinical 18F-FDG and amyloid brain positron emission tomography/CT in the investigation of cognitive impairment: Where are we now? Br. J. Radiol. 2019, 92, 20181027. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Shanes, E.D.; McCord, M.; Reish, N.J.; Flanagan, M.E.; Mesulam, M.M.; Jamshidi, P. Neuropathology of Anti-Amyloid-β Immunotherapy: A Case Report. J. Alzheimers Dis. 2023, 93, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Lista, S.; Teipel, S.J.; Garaci, F.; Nisticò, R.; Blennow, K.; Zetterberg, H.; Bertram, L.; Duyckaerts, C.; Bakardjian, H.; et al. Perspective on future role of biological markers in clinical therapy trials of Alzheimer’s disease: A long-range point of view beyond 2020. Biochem. Pharmacol. 2014, 88, 426–449. [Google Scholar] [CrossRef] [PubMed]
- Nobili, F.; Arbizu, J.; Bouwman, F.; Drzezga, A.; Agosta, F.; Nestor, P.; Walker, Z.; Boccardi, M. EANM-EAN Task Force for the Prescription of FDG-PET for Dementing Neurodegenerative Disorders. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain 18F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur. J. Neurol. 2018, 25, 1201–1217. [Google Scholar] [CrossRef]
- Khosravi, M.; Peter, J.; Wintering, N.A.; Serruya, M.; Shamchi, S.P.; Werner, T.J.; Alavi, A.; Newberg, A.B. 18F-FDG Is a Superior Indicator of Cognitive Performance Compared to 18F-Florbetapir in Alzheimer’s Disease and Mild Cognitive Impairment Evaluation: A Global Quantitative Analysis. J. Alzheimers Dis. 2019, 70, 1197–1207. [Google Scholar] [CrossRef]


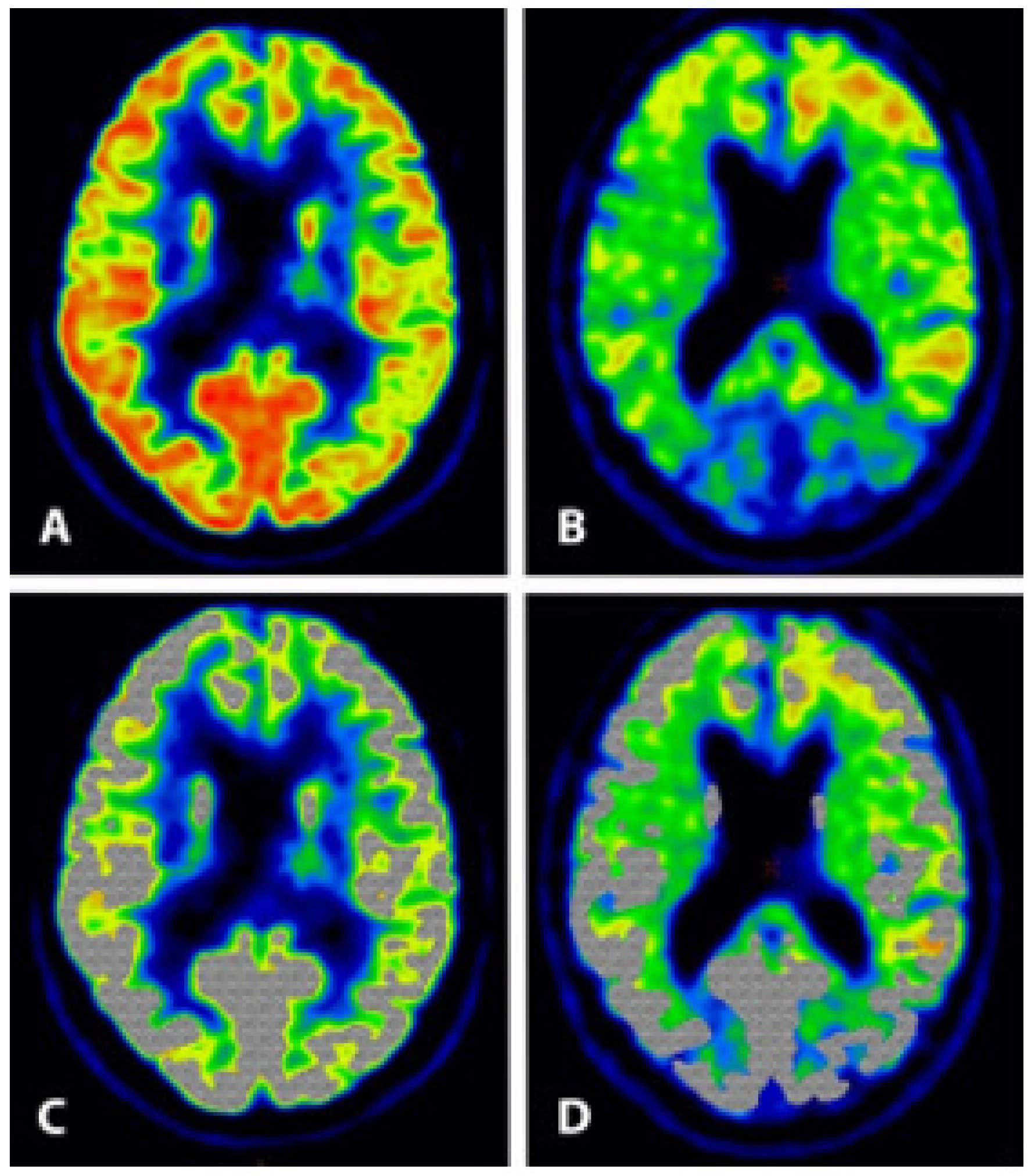{kind=link}
| Biomarker Group | Biomarkers |
|---|---|
| A (Aβ plaques) | Cortical amyloid-PET binding Low CSF Aβ42 Low Aβ42/Aβ40 ratio |
| T (Tau) | CSF phosphorylated tau (P-tau) Tau-PET |
| N (Neurodegeneration/-injury) | Anatomic MRI FDG-PET CSF total tau |
| Drug, Author, Year, Trial Name | Subjetcs | Change on Clinical Scale T vs. P | Amyloid Change | ARIA-E T vs. P | ARIA-H T vs. P | Volume Loss |
|---|---|---|---|---|---|---|
| Aducanumab Sevigny et al., 2016 [64] PRIME | Prodromal or mild AD Positive Aβ PET 3 T vs. 1 P group | CDR-SB (0–18): 0.7 vs. 1.8 MMSE (0–30): −0.6 vs. −2.8 | −27% | 41% vs. 0% | 9% vs. 5% | NR |
| Aducanumab Budd Haeberlein et al., 2022 [6], EMERGE | MCI due to AD or mild AD dementia Amyloid positivity | CDR-SB (0–18): 1.3 vs. 1.7 MMSE (0–30): −2.7 vs. −3.3 | −71% | 35%/26% vs. 2% | 20%/16% vs. 7% | Yes |
| Aducanumab Budd Haeberlein et al., 2022 [6], ENGAGE | MCI due to AD or mild AD dementia Amyloid positivity | CDR-SB (0–18): 1.6 vs. 1.6 MMSE (0–30): −3.6 vs. −3.6 | −58% | 36%/26% vs. 3% | 19%/16% vs. 6% | Yes |
| Lecanemab Swanson et al., 2021 [66] BAN2401-G000–201 | MCI or mild AD Amyloid | ADCOMS (0–1.97): −0.14 vs. −0.18 CDR-SB (0–18): 1.1 vs. 1.5 | −31% | 9.9% vs. 0.8% | 6.8% vs. 5.3% | Yes |
| Lecanemab Van Dyck et al., 2023 [9] Clarity-AD | MCI or mild AD Amyloid | CDR-SB (0–18): 1.2 vs. 1.7 ADCOMS (0–1.97): −0.16 vs. −0.22 | −55% | 12.6% vs. 1.7% | 17.3% vs. 9.0% | NR |
| Donanemab Mintun et al., 2021 [11] TRAILBLAZER-ALZ | Prodromal AD (MCI incl.) and mild AD Amyloid positivity | iADRS (0–144): −6.9 vs. −10.1 CDR-SB (0–18): 1.2 vs. 1.6 | −85% | 26.7% vs. 0.8% | 8.4% vs. 3.2% | NR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Høilund-Carlsen, P.F.; Revheim, M.-E.; Costa, T.; Kepp, K.P.; Castellani, R.J.; Perry, G.; Alavi, A.; Barrio, J.R. FDG-PET versus Amyloid-PET Imaging for Diagnosis and Response Evaluation in Alzheimer’s Disease: Benefits and Pitfalls. Diagnostics 2023, 13, 2254. https://doi.org/10.3390/diagnostics13132254
Høilund-Carlsen PF, Revheim M-E, Costa T, Kepp KP, Castellani RJ, Perry G, Alavi A, Barrio JR. FDG-PET versus Amyloid-PET Imaging for Diagnosis and Response Evaluation in Alzheimer’s Disease: Benefits and Pitfalls. Diagnostics. 2023; 13(13):2254. https://doi.org/10.3390/diagnostics13132254
Chicago/Turabian StyleHøilund-Carlsen, Poul F., Mona-Elisabeth Revheim, Tommaso Costa, Kasper P. Kepp, Rudolph J. Castellani, George Perry, Abass Alavi, and Jorge R. Barrio. 2023. "FDG-PET versus Amyloid-PET Imaging for Diagnosis and Response Evaluation in Alzheimer’s Disease: Benefits and Pitfalls" Diagnostics 13, no. 13: 2254. https://doi.org/10.3390/diagnostics13132254
APA StyleHøilund-Carlsen, P. F., Revheim, M.-E., Costa, T., Kepp, K. P., Castellani, R. J., Perry, G., Alavi, A., & Barrio, J. R. (2023). FDG-PET versus Amyloid-PET Imaging for Diagnosis and Response Evaluation in Alzheimer’s Disease: Benefits and Pitfalls. Diagnostics, 13(13), 2254. https://doi.org/10.3390/diagnostics13132254