
When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms

Abstract
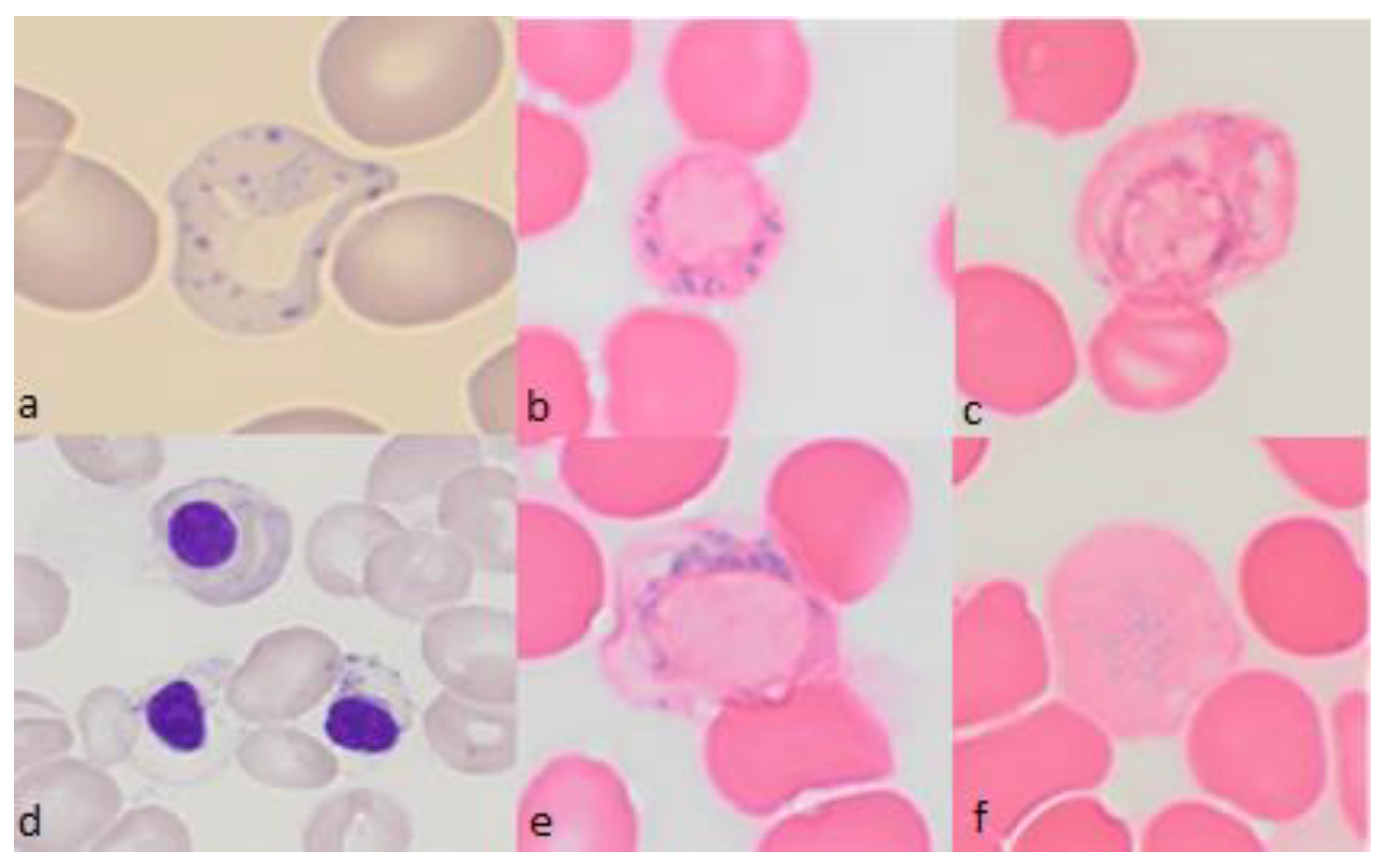
1. Introduction
2. Diagnosis
3. The Different Forms of Sideroblastic Anemia
3.1. Congenital Sideroblastic Anemias
3.1.1. Non-Syndromic Forms
3.1.2. Syndromic Forms
3.2. Secondary Acquired Sideroblastic Anemias
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mc, F.A.; Davis, L.J. Iron-staining erythrocytic inclusions with especial reference to acquired haemolytic anaemia. Glasg. Med. J. 1947, 28, 237–279. [Google Scholar]
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellstrom-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717. [Google Scholar] [CrossRef]
- Cazzola, M.; Invernizzi, R. Ring sideroblasts and sideroblastic anemias. Haematologica 2011, 96, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Ineffective erythropoiesis and its treatment. Blood 2022, 139, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Introduction to a How I Treat series on acquired hemolytic anemia. Blood 2021, 137, 1269. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO classification of tumors of haematopoeitic and lymphoid tissues. In WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; IARC: Lyon, France, 2017; pp. 98–106. [Google Scholar]
- Long, B.; Shi, H.; Zhu, C. Clinical analysis and literature review of a case with the myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Hematology 2020, 25, 283–285. [Google Scholar] [CrossRef]
- Nathan, D.I.; Feld, J.; El Jamal, S.M.; Mascarenhas, J.; Tremblay, D. Myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis: Ringing in a new future. Leuk. Res. 2022, 115, 106820. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Steensma, D.P. Dysplasia has A differential diagnosis: Distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr. Hematol. Malig. Rep. 2012, 7, 310–320. [Google Scholar] [CrossRef]
- Solis, M.; Perrin, J.; Guedenet, J.C.; Lesesve, J.F. RBCs inclusions after splenectomy: Not only Howell-Jolly bodies! Ann. Biol. Clin. 2013, 71, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, G. Transient appearance of ring sideroblasts in peripheral blood in the acute phase of secondary hemolytic anemia. Blood 2019, 133, 1000. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J. Clin. Investig. 2020, 130, 5245–5256. [Google Scholar] [CrossRef]
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69. [Google Scholar] [CrossRef]
- Long, Z.; Li, H.; Du, Y.; Han, B. Congenital sideroblastic anemia: Advances in gene mutations and pathophysiology. Gene 2018, 668, 182–189. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Richardson, D.R.; Prchal, J.; Ponka, P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol. 2009, 122, 120–133. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)–“2021 update on diagnosis, risk-stratification, and management”. Am. J. Hematol. 2021, 96, 379–394. [Google Scholar] [CrossRef]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Cattivelli, K.; Campagna, D.R.; Schmitz-Abe, K.; Heeney, M.M.; Yaish, H.M.; Caruso Brown, A.E.; Kearney, S.; Walkovich, K.; Markianos, K.; Fleming, M.D.; et al. Ringed sideroblasts in beta-thalassemia. Pediatr. Blood Cancer 2017, 64, e26324. [Google Scholar] [CrossRef]
- Hij, A.; Meunier, A.S.; Vignon, G.; Labrousse, J.; Augereau, P.F.; Carrere, F.; Aucher, P.; Lellouche, F. Rare sideroblastic anemias: About 2 cases, review of the literature and reminder of the main etiologies. Ann. Biol. Clin. 2021, 79, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Falcon, C.P.; Howard, T.H. An infant with Pearson syndrome: A rare cause of congenital sideroblastic anemia and bone marrow failure. Blood 2017, 129, 2710. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Wiseman, D.H.; May, A.; Jolles, S.; Connor, P.; Powell, C.; Heeney, M.M.; Giardina, P.J.; Klaassen, R.J.; Chakraborty, P.; Geraghty, M.T.; et al. A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood 2013, 122, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Schmitz-Abe, K.; Kennedy, E.K.; Mamady, H.; Naas, T.; Durie, D.; Campagna, D.R.; Lau, A.; Sendamarai, A.K.; Wiseman, D.H.; et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood 2014, 124, 2867–2871. [Google Scholar] [CrossRef]
- Faraji-Goodarzi, M.; Tarhani, F.; Taee, N. Dyserythropoiesis and myelodysplasia in thiamine-responsive megaloblastic anemia syndrome. Clin. Case Rep. 2020, 8, 991–994. [Google Scholar] [CrossRef]
- Natelson, E.A.; Pyatt, D. Acquired myelodysplasia or myelodysplastic syndrome: Clearing the fog. Adv. Hematol. 2013, 2013, 309637. [Google Scholar] [CrossRef][Green Version]
- Delmotte, V.; Foidart, P.; De, V.A.; Lejeune, M.; Delwaide, J. Diagnosis of anemia associated with alcoholic cirrhosis. Rev. Med. Liege 2019, 74, 527–534. [Google Scholar]
- Pol, R.R.; Howard, M.R. A man with anemia and a change in personality. Blood 2014, 123, 1784. [Google Scholar] [CrossRef]
- Colucci, G.; Silzle, T.; Solenthaler, M. Pyrazinamide-induced sideroblastic anemia. Am. J. Hematol. 2012, 87, 305. [Google Scholar] [CrossRef]
- Saini, N.; Jacobson, J.O.; Jha, S.; Saini, V.; Weinger, R. The perils of not digging deep enough—Uncovering a rare cause of acquired anemia. Am. J. Hematol. 2012, 87, 413–416. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Willekens, C.; Dumezy, F.; Boyer, T.; Renneville, A.; Rossignol, J.; Berthon, C.; Cotteau-Leroy, A.; Mehiaoui, L.; Quesnel, B.; Preudhomme, C. Linezolid induces ring sideroblasts. Haematologica 2013, 98, e138–e140. [Google Scholar] [CrossRef] [PubMed]
- Liapis, K.; Vrachiolias, G.; Spanoudakis, E.; Kotsianidis, I. Vacuolation of early erythroblasts with ring sideroblasts: A clue to the diagnosis of linezolid toxicity. Br. J. Haematol. 2020, 190, 809. [Google Scholar] [CrossRef] [PubMed]
- Vial, T.; Grignon, M.; Daumont, M.; Guy, C.; Zenut, M.; Germain, M.L.; Jaubert, J.; Ruivard, M.; Guyotat, D.; Descotes, J. Sideroblastic anaemia during fusidic acid treatment. Eur. J. Haematol. 2004, 72, 358–360. [Google Scholar] [CrossRef]
- Ok, C.Y.; Medeiros, L.J.; Hu, Y.; Bueso-Ramos, C.E.; Wang, S.A. Transient/reversible ring sideroblasts in bone marrow of patients post cytotoxic therapies for primary malignancies. Leuk. Res. 2011, 35, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Ramlal, R. “Myelodysplasia” from copper deficiency. Blood 2019, 133, 883. [Google Scholar] [CrossRef]
- Villalba, A.; Senent, L. Differential diagnosis of myelodysplastic syndrome: Anemia associated with copper deficiency. Blood 2018, 131, 1389. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. New drugs for myeloid neoplasms with ring sideroblasts: Luspatercept vs imetelstat. Am. J. Hematol. 2021, 96, 761–763. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Platzbecker, U.; Fenaux, P.; Zeidan, A.M.; Garcia-Manero, G.; Mufti, G.J.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Jurcic, J.G.; et al. Luspatercept for myelodysplastic syndromes/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Leukemia 2022, 36, 1432–1435. [Google Scholar] [CrossRef]
- Jouzier, C.; Cherait, A.; Cony-Makhoul, P.; Hamel, J.F.; Veloso, M.; Thepot, S.; Cluzeau, T.; Stamatoullas, A.; Garnier, A.; Guerci-Bresler, A.; et al. Red blood cell transfusion burden in myelodysplastic syndromes (MDS) with ring Sideroblasts (RS): A retrospective multicenter study by the Groupe Francophone des Myelodysplasies (GFM). Transfusion 2022, 62, 961–973. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jadersten, M.; Jansson, M.; Elena, C.; Galli, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zeinah, G.; DeSancho, M.T. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J. Blood Med. 2020, 11, 305–318. [Google Scholar] [CrossRef] [PubMed]






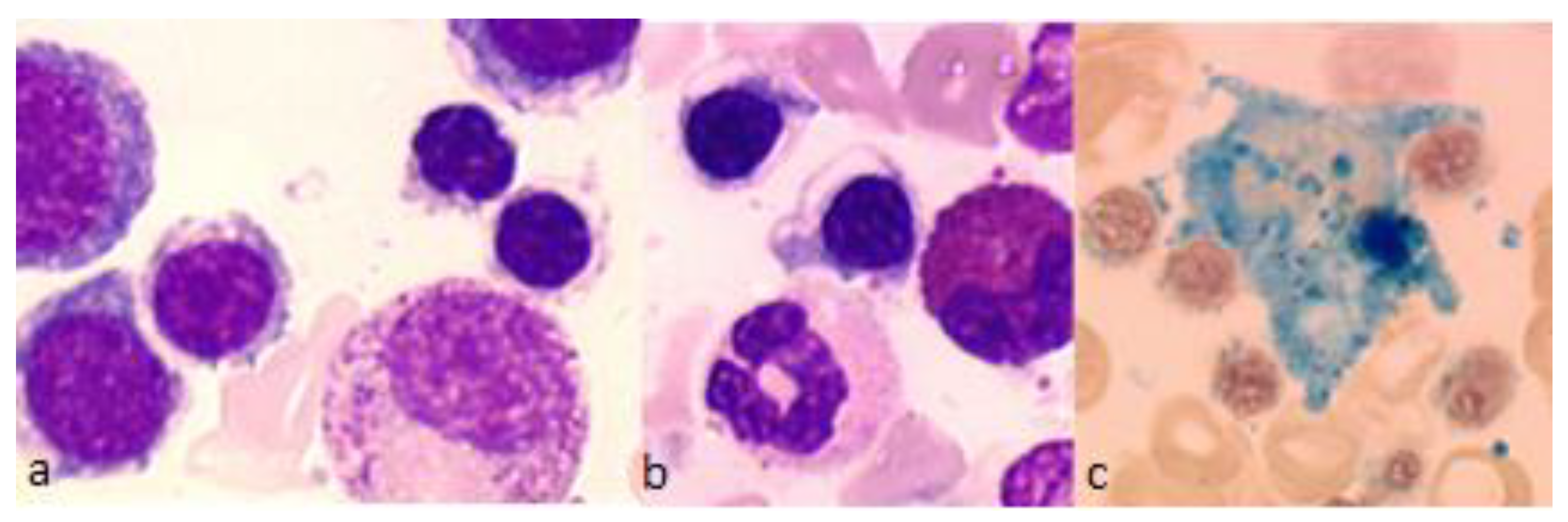{kind=link}
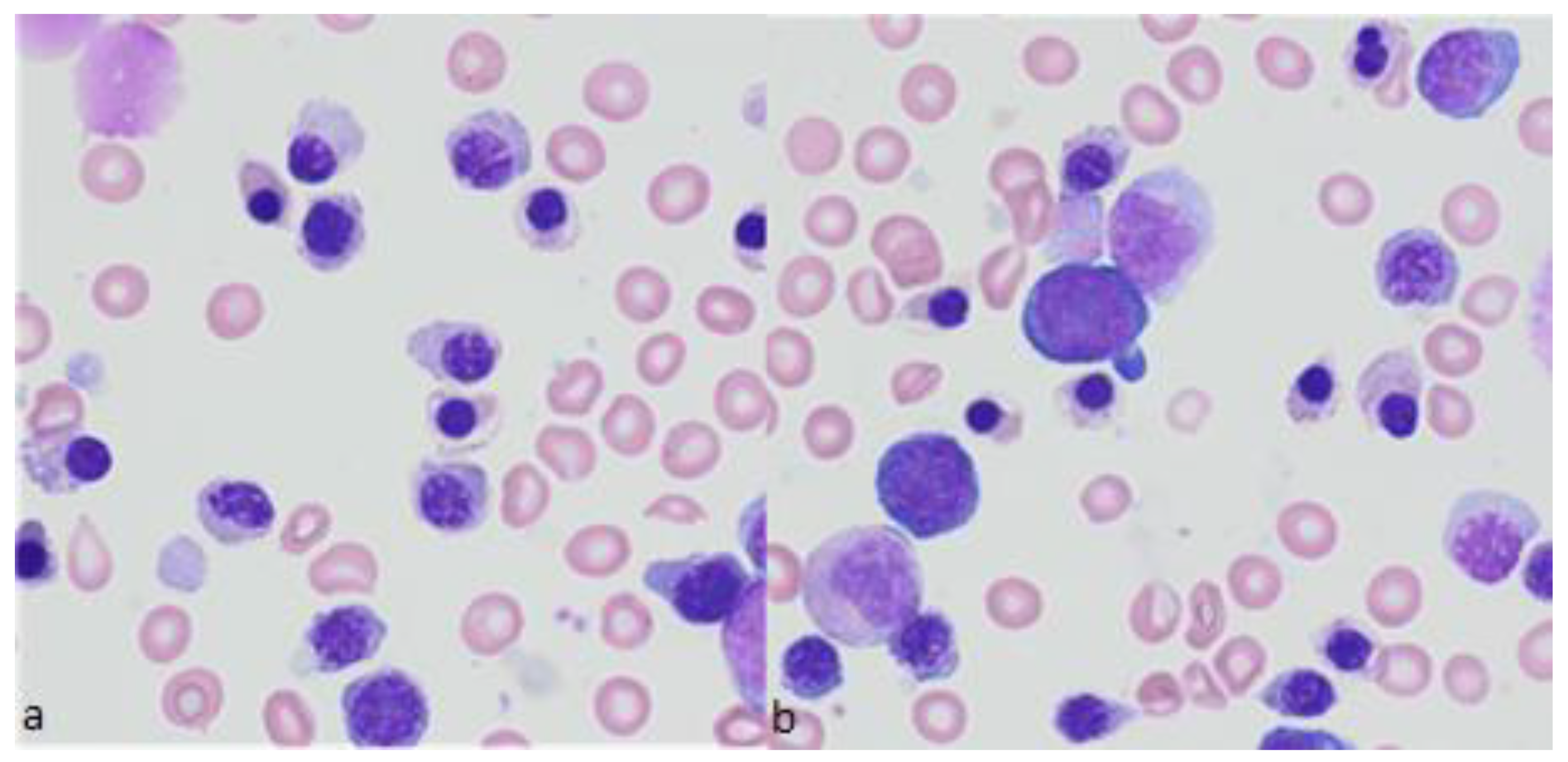{kind=link}
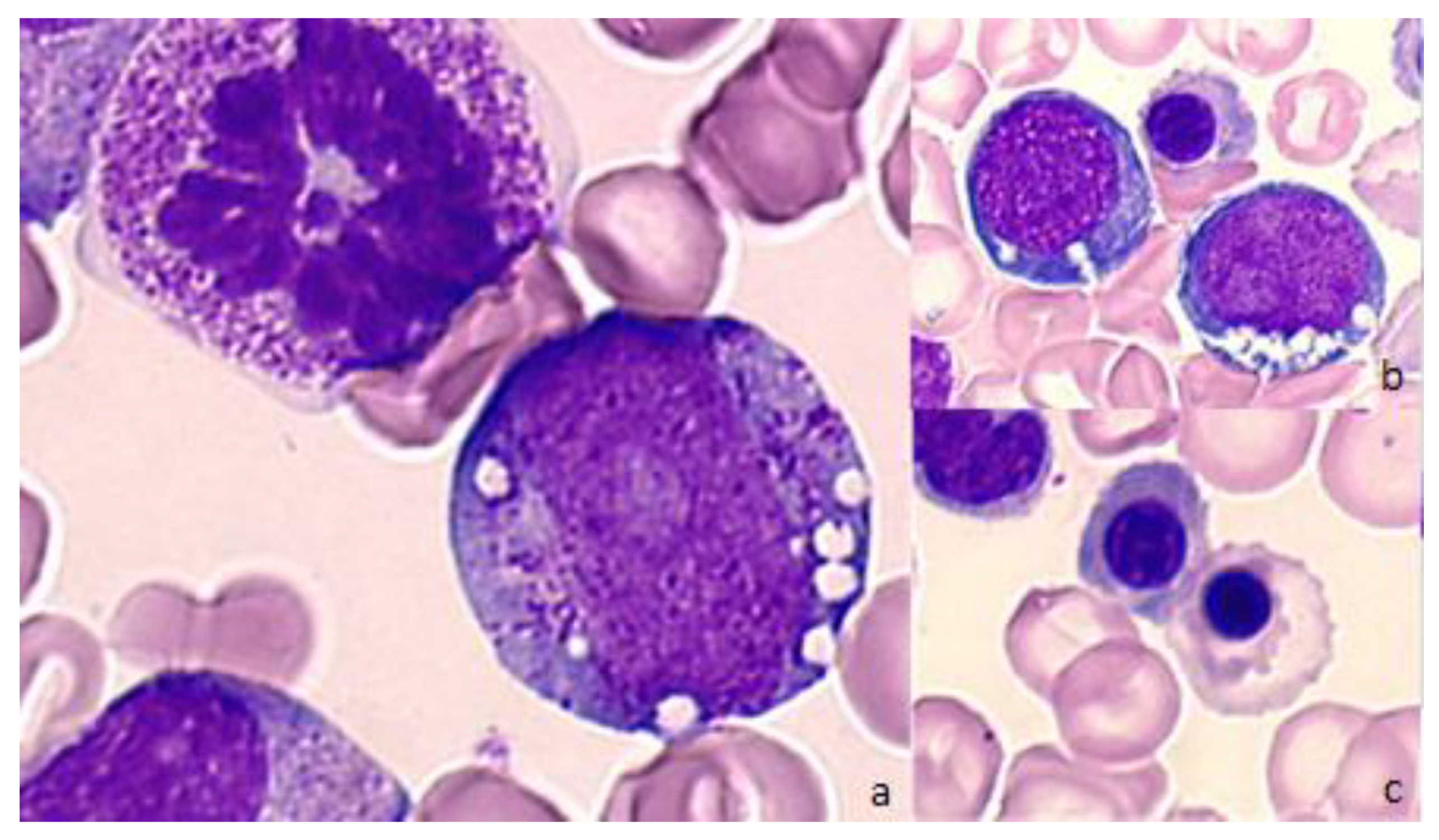{kind=link}
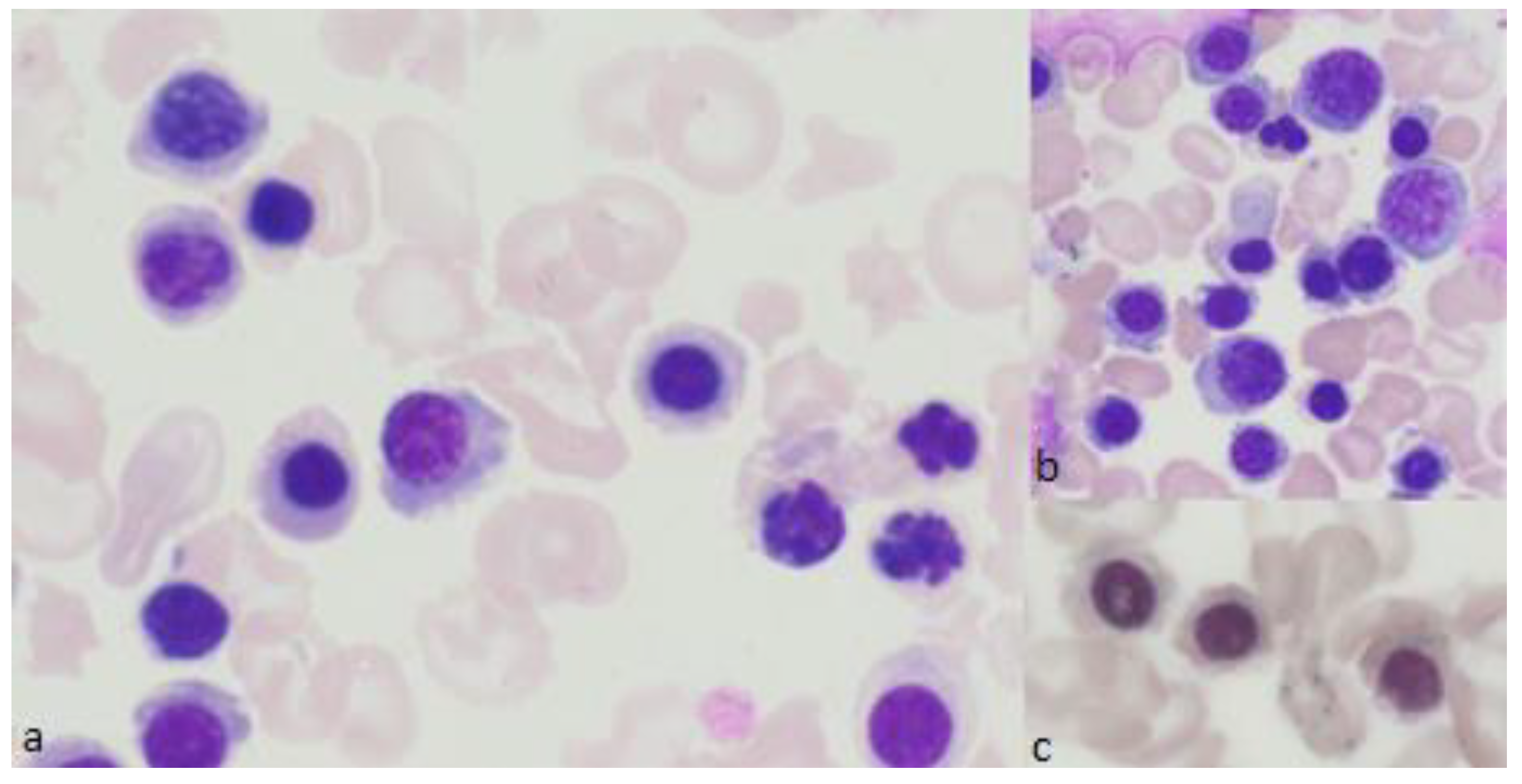{kind=link}
{kind=link}
| Condition | Inheritance | Mutations | Biological Features |
|---|---|---|---|
| MDS with low blasts and SF3B1 mutation/MDS with low blasts and ring sideroblasts (MDS-RS: 2016 revised WHO) | Somatic | SF3B1 | Cytopenia of one or two lineages Mild to severe isolated anemia Normal or high mean corpuscular volume Morphological single or multilineage dysplasia <2% blasts in peripheral blood <5% blasts in bone marrow (no Auer rods) Perls’ stain: ≥15% RS with wild-type SF3B1 or ≥5% RS in the presence of SF3B1 mutation |
| MDS/MPN with SF3B1 mutation and thrombocytosis (MDS/MPN -RS-T: 2016 revised WHO) | Somatic | SF3B1 +/− JAK2 V617F | Persistent thrombocytosis (≥450 giga/L) Megakaryocytic hyperplasia and morphological abnormalities MPN like Moderate anemia with erythroid lineage dysplasia, with or without multilineage dysplasia Normal or high mean corpuscular volume <1% blasts in peripheral blood <5% blasts in bone marrow Perls’ stain: ≥15% RS with or without SF3B1 mutation |
| Condition | Gene Anomaly/Chromosomal Localization |
|---|---|
| Non-syndromic sideroblastic anemias | |
| Heme synthesis defects | |
| XLSA = X-linked sideroblastic anemia | ALA synthase gene (ALAS2)—Xp11.21 |
| SIDBA2 = Autosomal recessive pyridoxine refractory sideroblastic anemia | SLC25A38—3p22.1 |
| EPP = Erythropoietic protoporphyria | FECH—18q22 |
| Fe-S biogenesis defects | |
| GLRX5 deficiency | GLRX5—14q32 |
| HSPA9 deficiency | HSPA9—5q31.2 |
| HSCB deficiency | HSCB—22q12.1 |
| Syndromic sideroblastic anemias | |
| Fe-S biogenesis defects | |
| XLSA/A = X-linked sideroblastic anemia and spinocerebellar ataxia | ABCB7—Xq13.1-q13.3 |
| Mitochondrial protein synthesis defects | |
| MLASA1 = Myopathy, lactic acidosis and sideroblastic anemia | YARS2—12p11.21 |
| MLASA2 = Myopathy, lactic acidosis and sideroblastic anemia | PUS1—12q24.33 |
| LARS2 deficiency | LARS2—3p21.3 |
| SFID syndrome | TRNT1—3p26.1 |
| Pearson syndrome | Mitochondrial DNA deletion |
| Mitochondrial respiratory protein mutations | |
| TRMA = Thiamine-responsive megaloblastic anemia | SLC19A2—1q23.3 |
| MT-ATP6-sideroblastic anemia | MT-ATP6 |
| NDUFB11—sideroblastic anemia | NDUFB11–X p11.23 |
| Acquired Reversible Sideroblastic Anemias | |
|---|---|
| Exposure to toxic substances | Alcoholism Heavy metal intoxication (lead, arsenic, mercury) Benzene exposure |
| Drugs | Anti-tuberculosis (isoniazid, pyrazinamide, cycloserine) Antibiotics (chloramphenicol D-penicillamine, linezolid, lincomycin, cefadroxil, fusidic acid, tetracyclines) Cancer chemotherapy (chlorambucil, busulfan, melphalan, lenalidomide) |
| Malnutrition/deficiency in nutrition or other metabolic disorders | Vitamin B1, B6, B9, and B12 deficiencies Copper deficiency Zinc overdose Prolonged parenteral nutrition Gastric surgery Hypothermia |
| Acquired clonal sideroblastic anemias = myelodysplastic neoplasms (or MDS/MPN neoplasms) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girard, S.; Genevieve, F.; Rault, E.; Fenneteau, O.; Lesesve, J.-F. When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics 2022, 12, 1752. https://doi.org/10.3390/diagnostics12071752
Girard S, Genevieve F, Rault E, Fenneteau O, Lesesve J-F. When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics. 2022; 12(7):1752. https://doi.org/10.3390/diagnostics12071752
Chicago/Turabian StyleGirard, Sandrine, Franck Genevieve, Emmanuelle Rault, Odile Fenneteau, and Jean-François Lesesve. 2022. "When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms" Diagnostics 12, no. 7: 1752. https://doi.org/10.3390/diagnostics12071752
APA StyleGirard, S., Genevieve, F., Rault, E., Fenneteau, O., & Lesesve, J.-F. (2022). When Ring Sideroblasts on Bone Marrow Smears Are Inconsistent with the Diagnosis of Myelodysplastic Neoplasms. Diagnostics, 12(7), 1752. https://doi.org/10.3390/diagnostics12071752