Abstract
Surfactant is a complex of phospholipids and proteins produced in type II pneumocytes. Its deficiency frequently occurs in preterm infants and causes respiratory distress syndrome. In full-term newborns, its absence results from mutations in the SFTPC, SFTPB, NKX2-1, or ABCA3 genes involved in the surfactant metabolism. ABCA3 encodes ATP-binding cassette, which is responsible for transporting phospholipids in type II pneumocytes. We present a case of a male late preterm newborn with inherited surfactant deficiency in whom we identified the likely pathogenic c.604G>A variant in one allele and splice region/intron variant c.4036-3C>G of uncertain significance in the second allele of ABCA3. These variants were observed in trans configuration. We discuss the diagnostic challenges and the management options. Although invasive treatment was introduced, only temporary improvement was observed. We want to raise awareness about congenital surfactant deficiency as a rare cause of respiratory failure in term newborns.
1. Introduction
Surfactant plays a crucial role in reducing surface tension in alveoli to maintain stable air space for gas exchange and prevent end-expiratory alveolar collapse. Its deficiency is mainly observed in premature neonates, as it may lead to respiratory distress syndrome. Congenital surfactant deficiency caused by mutations in various genes playing an important role in surfactant biosynthesis may cause a similar presentation of severe respiratory distress syndrome with lethal respiratory failure in full-term newborns or interstitial lung disease (ILD) in older children and adults [1,2]. Congenital surfactant deficiency is associated with mutations in the SFTPC, SFTPB, NKX2-1, or ABCA3 as well as CSF2RA, CSF2RB, SFTPA1, SFTPA2, SFTA3, and SFTPD genes involved in the surfactant biosynthesis [3,4].
Surfactant is a complex of highly specific phospholipids and proteins synthesized by epithelial cells called type II pneumocytes. It is intracellularly embedded in inclusion organelles-lamellar bodies. Phospholipids make up almost 90% of surfactant weight; the remaining 10% are hydrophobic proteins SP-B and SP-C and hydrophilic proteins from the collectin family SP-A and SP-D [5]. The specific surfactant proteins A (SP-A), B (SP-B), C (SP-C), and D (SP-D) are encoded by the SFTPA, SFTPB, SFTPC, and SFTPD genes and have considerable functional significance [6]. Type II pneumocytes differentiate between weeks 24 and 34 of gestation. Premature newborns with RDS have only about one-tenth the amount of surfactant compared to healthy full-term newborns [6]. Mutations of SFTPB, SFTPC, and ABCA3 genes can lead to qualitative and quantitative surfactant defects [2]. However, the most common cause of primary surfactant defect is the loss of function mutations in the ABCA3 gene [7].
ABCA3 is a member of the ATP-binding cassette (ABC) transporter family, which encodes membrane proteins that transport compounds across biologic membranes. It is a 1704-amino-acid protein expressed mainly in the alveolar epithelial cells at the limiting membrane of lamellar bodies [7]. It has been detected from 26–27 weeks of gestation in normal fetuses [4]. The ABCA3 is responsible for transporting phospholipids into the lamellar bodies (LBs) in type II pneumocytes. It is crucial for AT2 cells’ homeostasis, lipid composition, and protein maturation of surfactant. There have already been more than 150 patients with recessive mutations identified in the ABCA3 gene that may lead to respiratory distress in full-term newborns and older children [8]. Bi-allelic ABCA3 mutations were the most frequent cause of congenital surfactant deficiency [6].
We report a case of a late preterm infant with severe respiratory failure from the first day of life, in whom we identified ABCA3 variants in trans c.604G>A and c.4036-3C>G. These variants were detected in the mother and father of the presented child in a heterozygous state. ABCA3 variants in both alleles may cause a loss of function of the ABCA3 protein and significant surfactant deficiency with severe respiratory failure.
2. Case Report
The patient was a late-preterm baby boy, delivered normally at the primary care center at 36 weeks of gestation, after an uncomplicated pregnancy, with a birth weight of 2097 g and Apgar scores of 10 in the 1st and the 5th minute of life. The parents do not have another child together. The mother has three healthy children with her previous partner.
Within the first few hours of his life, he developed severe respiratory distress and required respiratory support. At first, the nCPAP therapy was introduced, and the patient was transported to the tertiary care center. After several hours he required non-invasive ventilation, and since his status continued to worsen, mechanical ventilation was initiated on the 3rd day of life. Ultimately, due to increasing respiratory acidosis, he was put on high-frequency ventilation (HFO) on the 5th day of life. He also required inhaled nitric oxide from the 4th to the 15th day of life and later from the 23rd until the 30th day of life. The baby received two doses of exogenous surfactant during hospitalization on the 6th and 21st day of life without significant improvement. The corticosteroids (dexamethasone) were administered twice in 10-day courses (0.03 mg/kg twice a day) without any progress. Echocardiography excluded congenital heart defects. The mechanical ventilation was complicated by pneumothorax on the 23rd day of life, treated with suction drainage for seven days.
On pulmonary ultrasound, bilaterally compact B-artifacts were observed, which created the image of white lungs. Chest radiograms showed diffuse opacification of both lungs with air bronchograms. Computed tomography showed a characteristic pattern of interstitial lung disease with ground-glass opacities and pneumatocele. In the extended screening test, cystic fibrosis was excluded. The patient was transferred to the otolaryngology department to broaden diagnostics. The bronchofiberoscopy raised suspicion of a large tracheoesophageal fistula, which was eventually excluded after the consecutive examination (Supplementary Video S1).
The patient was admitted to the Department of Newborns’ Infectious Diseases on the 54th day of life. He was on HFO ventilation, FiO2 100%. We performed the procedure of surfactant lung lavage as a rescue treatment, after which we observed transitional improvement, which enabled us to use conventional mechanical ventilation (SIMV) instead of HFO. However, FiO2 requirements and ventilator parameters remained very high, with oxygen demand of 1.0 and MAP of 16 cm H2O. On the 74th day of life, the patient presented with cardiac arrest after accidental extubation. He was resuscitated after 4 min of chest compressions and drugs administration.
The pulmonary ultrasound and chest radiographs performed frequently during the patient’s hospital stay did not change over time. Figure 1 presents pulmonary ultrasound on the 83rd day of life in which compact B-artifacts create the image of white lungs with many subpleural consolidations. The chest radiographs shown in Figure 2 are taken in exactly one month. On both X-rays, we can see diffuse reticular granularity and air bronchograms. Computed tomography repeated after several weeks (Figure 3 and Supplementary Video S2) showed again a pattern of interstitial lung disease with ground-glass opacities and pneumatocele.
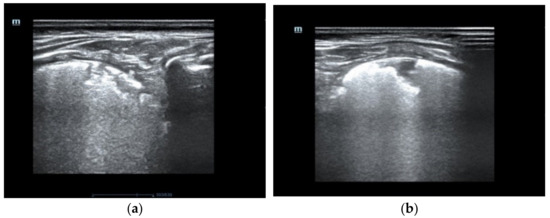
Figure 1.
The pulmonary ultrasound shows compact B artifacts creating the image of white lungs with many subpleural consolidations. (a) posterior part of the right lung (b) the rear part of the left lung.
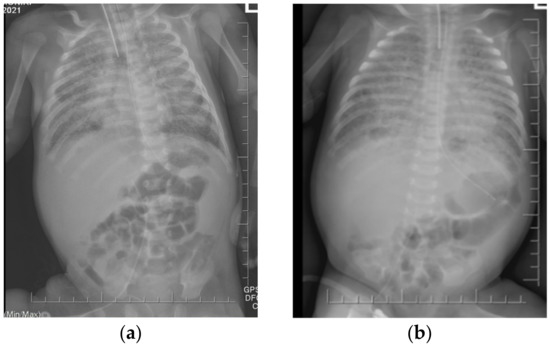
Figure 2.
The chest X-rays revealed diffuse opacification of both lungs with air bronchograms. No improvement has been observed during one month. (a) 54th day of life (b) 85th day of life.
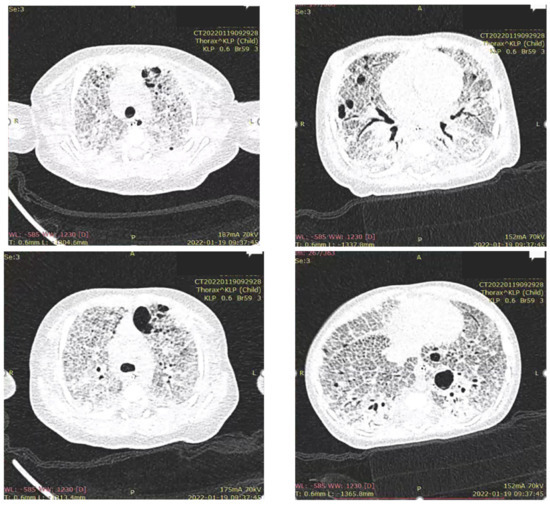
Figure 3.
Computed tomography shows a characteristic pattern of interstitial lung disease with ground-glass opacities and pneumatocele.
In microbial culture from the bronchotracheal aspirate on the 67th day of life, Serratia marcescens was found. At the same time, profuse discharge from the airways was observed, and CRP was elevated and rising (from 11.72 up to 81.05 mg/L), so we diagnosed ventilator-associated pneumonia. The patient was treated with antibiotics for 21 days, but it did not cause any improvement in his general condition, although the inflammation markers returned to typical values.
Due to a very unstable state, the patient was given high doses of sedative drugs (Morphine 50 µg/kg/h and Midazolam 0.7 mg/kg/h). This strategy allowed us to change respiration parameters from SIMV with PIP approx. 40 cm H2O to PRVC with PIP approximately 30 cm H2O and PEEP of 12–14 cm H2O. We aimed to provide better comfort to the patient, so NAVA ventilation was introduced, which allowed lowering respiratory parameters, CO2 retention, and oxygen demand from 1.0 to 0.65–0.8 FiO2. The ventilator parameters on PRVC and NAVA ventilation are presented in Figure 4.
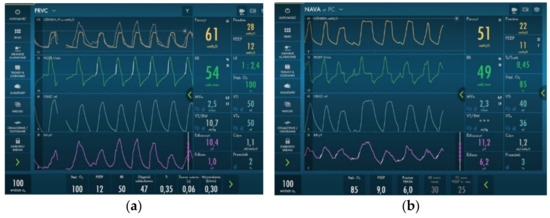
Figure 4.
Invasive ventilation parameters used during hospitalization. We observed a reduction in PIP and MAP parameters after introducing NAVA ventilation. (a) PRVC ventilation; (b) NAVA ventilation.
Genetic analysis of genes associated with surfactant metabolism disorders was performed during the hospital stay using next-generation sequencing. Informed consent was obtained from parents according to the local regulations. Genomic DNA was extracted from a peripheral blood EDTA sample. The most important genes involved in surfactant metabolism: ABCA3, SFTPB, and SFTPC, were analyzed with twist Bioscience Custom Panel in NextSeq Ilumina. No mutation was detected in the SFTPC and SFTPB genes; however, we identified the likely pathogenic c.604G>A variant in one allele and splice region/intron variant c.4036-3C>G of uncertain significance in the second allele of ABCA3. These variants were observed in trans configuration. Both parents are carriers of one variant each.
After the genetic result of the ABCA3 variant related to surfactant dysfunction and in the absence of the possibility of causal treatment of the disease, the patient was transferred to the hospital closer to his family to introduce palliative care. Following discussion with parents, the “do not resuscitate” form was signed by specialists. The patient deceased at the age of 99 days.
3. Discussion
In term newborns, respiratory distress occurs in 5% to 7% of live births, mainly resulting from the abnormal transition from fetal to neonatal life. It is primarily manifested by tachypnea, nasal flaring, intercostal or subcostal retractions, audible grunting, and cyanosis. Most cases are mild and transient and are diagnosed as transient tachypnea of the newborn (TTN). Among other common causes, we distinguish persistent pulmonary hypertension of newborns (PPHN), meconium aspiration syndrome (MAS), and infectious conditions such as sepsis or pneumonia. However, severe respiratory distress is often due to non-pulmonary causes like congenital heart disease, air leaks, or pathoanatomic conditions of the pulmonary airway. Occasionally, term neonatal respiratory distress is associated with an inherited primary lung disease such as surfactant metabolism defects [9,10].
The ABCA3 is a transmembrane protein found in pneumocytes type II lamellar bodies. It belongs to a more incredible family of ABC transporters, which mediate the transport of various physiologic lipid compounds. Mutations of their encoding genes are linked to several diseases, e.g., Stargardt’s disease (ABCA4), harlequin ichthyosis (ABCA12) [11]. ABCA3 is a protein encoded by an 80 kb gene located on the 16th chromosome (16p13.3). The incidence of ABCA3 mutation may vary between 1:4400 to 1:20,000 in the European and African populations [12].
ABCA3 deficiency is one of the most frequent causes of genetic surfactant metabolism disorders [11] and should be suspected in neonates with severe neonatal respiratory distress syndrome refractory to conventional treatment [8]. It leads to abnormal lamellar bodies’ structure and abnormal surfactant lipid composition, associated with impaired processing of SP-B and SP-C proteins [13]. Surfactant isolated from infants with mutations in ABCA3 presents decreased function and deficiency in phosphatidylcholine [14].
Qualitative functional characterization of ABCA3 missense variants suggests 2 pathogenic classes: disrupted intracellular trafficking (type I mutant) or impaired ATPase-mediated phospholipid transport into the lamellar bodies (type II mutant). Each disease-associated ABCA3 variant is associated with a diverse pulmonary phenotype [15].
The loss of ABCA3 function leads to dramatic progress of neonatal distress syndrome and death within the first three months of life, whereas residual function is associated with milder disease [8]. The most common presentation of a baby with an ABCA3 mutation that leads to respiratory failure is a full-term baby with moderate to severe respiratory distress and signs of diffuse lung disease without satisfactory history or laboratory findings [2,16]. The variants in ABCA3 are unique to individuals and families and have a homozygous or compound heterozygous state. Heterozygous ABCA3 missense variants are present in 1.5–3.7% of African and European descent individuals [12]. The most frequent mutation of the ABCA3 gene in Europe is p. Glu292Val. It is carried by 1.3% of the Danish population [17]. This mutation is responsible for less than 10% of identified pathogenic alleles [18]. Most mutations are unique, with over 300 different variants reported in the literature, making genetic counseling difficult in the families, especially in the case of missense mutations (amino acid substitution) where phenotype-genotype correlation is unknown. In our patient, SFTPB, SFTPC, and ABCA3 genes associated with congenital surfactant deficiency were analyzed by next-generation sequencing. Two variants were identified: likely pathogenic missense variant: c.604G>A (p.Gly202Arg) and splice region/intron variant c.4036-3C>G. The first variant has already been described in a patient with congenital surfactant deficiency in Iran [19]. The other one was not defined yet, but this variant can disturb RNA splicing resulting in the loss of exons or the inclusion of introns and an altered protein-coding sequence. Identification of homozygous or compound heterozygous ABCA3 gene mutations, commonly predicts more challenging clinical profiles and poor outcomes compared to patients with a single ABCA3 mutation and those with no defined genetic abnormalities [20], which is in agreement with the severe condition and lethal outcome of our patient.
The histopathological findings in the lungs of the patients with congenital surfactant deficiency consist of AEC2 hyperplasia, pulmonary alveolar proteinosis (PAP), inter-alveolar septa thickening, fibrosis, and inflammation. Frequently, prominent foamy macrophages are found in the airspaces, often embedded in proteinaceous material. Although the findings of routine examination may indicate one of the disorders, they cannot distinguish among different genetic causes since they are nonspecific for any of the Sp-B, Sp-C, and ABCA3 mutations [21]. A molecular diagnosis is needed to determine the specific mutation affecting each case. Therefore, the histopathological examination was not performed in our patient’s case.
In the differential diagnosis of unclear neonatal respiratory failure, we need to take into consideration also other rare causes of respiratory distress. Among them are lethal lung developmental disorders, which result from abnormal or suppressed lung development during the fetal period. In this group, we distinguish alveolar capillary dysplasia with misalignment of the pulmonary veins (ACDMPV), acinar dysplasia (AcDys), congenital alveolar dysplasia (CAD), and pulmonary hypoplasia (PH) [10].
Another rare cause of neonatal respiratory failure might be congenital central hypoventilation syndrome, also known as Ondine’s Curse, caused by a mutation in the PHOX2B gene and characterized by severely impaired central autonomic control of breathing and dysfunction of the autonomous nervous system [10,22].
Apart from pulmonary causes, it is crucial to exclude any otolaryngeal congenital disabilities. In our patient, for some time, the tracheoesophageal fistula was suspected. Bronchoscopy suggested the presence of a large canal on the posterior tracheal wall. Additionally, the mucous membrane of the trachea was macerated, and many thrombi were found in the respiratory tract, which suggested destruction due to aspiration through the fistula. After administering total parenteral nutrition, we observed a recovery of the mucous membrane, which was first interpreted as a result of proper fistula management. Since the diagnosis was eventually ruled out, it seems that the injury of the mucous membrane was due to aggressive ventilation with very high ventilatory pressure, and it improved only when we managed to stabilize the patient and reduce the respiratory parameters. Throughout the diagnostic process in newborns with unknown causes of severe respiratory distress, it is crucial to be careful with the interpretation of endoscopic imagination. The worsening of the mucous membrane of the respiratory tract might result from various conditions, both anatomic or pathophysiologic.
It is also worth mentioning that we observed a lack of ventilation reserve in our patient, leading to fast heart arrest when extubation occurred.
As of today, there is no causal treatment for congenital surfactant deficiency. The starting point remains the management of respiratory distress by ventilation and oxygen therapy. Anti-inflammatory therapy with steroid course was described by Ciantelli M. et al. to be unbeneficial [23]. Our patient also received two courses of steroids, and they did not cause any positive effects. Nishida D. et al. reported an improvement after azithromycin administration, although the exact mechanism of this therapy is still unclear [24]. Some cases report improvement in patients with interstitial lung disease after administration of hydroxychloroquine [25,26]. We did not try any of these therapies on our patient. The only transient positive effect that we observed was after lung lavage with surfactant. It enabled us to reduce the ventilation parameters and finish HFO ventilation. Similar short-time results after whole lung lavages were also described in the literature [27]. On the contrary, Si et al. reported favorable clinical outcomes in 3 infants with neonatal respiratory distress and compound heterozygous variants in ABCA3 treated with a 3-drug therapeutic regimen of monthly methylprednisolone pulses and daily azithromycin and hydroxychloroquine [28]. However, it is unknown whether the improvement was attributable to the medical regimen or the natural history of the lung disease. Further studies should focus on individualized, precision-based therapeutic regimens for patients with ABCA3 deficiency.
For now, it seems that a lung transplant is the only definitive treatment for most infants with severe congenital surfactant deficiency [29]. It remains the only option for end-stage lung disease. Its 5-year survival was described to be approximately 50% [30]. Unfortunately, there is poor availability of lung transfer in such young patients, and the number of centers performing these surgeries is limited due to logistic problems and the complexity of these interventions. Many patients need to be gathered to gain enough technical expertise while finding donors requires a dedicated network and program. Moreover, surgical and medical issues, like managing immunosuppression and follow-up in infants, make lung and cardiac transplants challenging to accomplish [31]. Although rarely performed in Europe, lung transplants have been undertaken at St Louis Children’s Hospital (St. Louis, MO, USA). Between 1993 and 2015, 44 children were reported; 28 were transplanted at age <1 year, and 16 were older [32].
Recently, Forstner et al. used a human pulmonary epithelial cell line (A549) and machine-learning algorithms to develop a phenotypic assay to detect morphologic differences between stably transfected cells expressing wild-type ABCA3 or missense variants. They next used this phenotypic assay to screen 1280 U.S. Food and Drug Administration (FDA)-approved small molecules and identified cyclosporine as a corrector of several ABCA3 mutants that disrupt intracellular trafficking [33]. Hence, cyclosporin A may be selected for orphan drug evaluation in controlled repurposing trials in patients with ABCA3 mutations [33,34].
Even though establishing the diagnosis does not alter the outcome of patients with congenital surfactant defects and frequently, the diagnosis is set after the patient’s death, it is essential to adequately counsel the parents and family members about the recurrence risk.
4. Conclusions
The patient’s clinical presentation is consistent with a case of respiratory failure due to surfactant deficiency. The histopathological findings in patients with surfactant deficiency are nonspecific for any SP-B, SP-C, and ABCA3 mutations, and a molecular diagnosis is needed. In the patient’s genetic analysis, two heterozygous in trans variants of the gene ABCA3 were found. We conclude that they are responsible for this patient’s respiratory failure. Genetic counseling was suggested for the parents and relatives, informed of the recurrence risks.
Congenital surfactant defects are rare conditions. They must be considered among term newborns with respiratory failure of unclear etiology. Many unknown, unique possible variants in several genes encoding proteins are crucial in surfactant metabolism. Therefore, it is strongly recommended to use simultaneous, broad-spectrum approaches such as next-generation sequencing with panels targeted to surfactant-related genes or whole-exome sequencing [8,10].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diagnostics12051084/s1, Video S1: Bronchoscopy, Video S2: CT scans.
Author Contributions
Conceptualization, K.W.-S. and T.S.; Writing—original draft preparation, J.R. and K.W.-S.; Writing—review and editing, K.W.-S.; Visualization, J.R. and K.W.-S.; Supervision, K.W.-S., T.S., R.Ś. and J.S.; Project administration, K.W.-S. and T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient’s parents to publish this paper.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Griese, M.; Haug, M.; Brasch, F.; Freihorst, A.; Lohse, P.; Von Kries, R.; Zimmermann, T.; Hartl, D. Incidence and classification of pediatric diffuse parenchymal lung diseases in Germany. Orphanet J. Rare Dis. 2009, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Malý, J.; Navrátilová, M.; Hornychová, H.; Looman, A.C. Respiratory failure in a term newborn due to compound heterozygous ABCA3 mutation: The case report of another lethal variant. J. Perinatol. 2014, 34, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, G.M.A.; Al Otaibi, W.H.; Al-Mobaireek, K.F.A.; Al-Saleh, S. Adenosine triphosphate-binding cassette member A3 gene mutation in children from one family from Saudi Arabia. Ann. Thorac. Med. 2016, 11, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Yang, P.; Wegner, D.J.; Heins, H.B.; Kaliberova, L.N.; Kaliberov, S.A.; Curiel, D.T.; White, F.V.; Hamvas, A.; Hackett, B.P.; et al. Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. Am. J. Respir. Cell Mol. Biol. 2016, 55, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Gower, W.A.; Nogee, L.M. Surfactant Dysfunction. Paediatr. Respir. Rev. 2011, 12, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Hallman, M. The Surfactant System Protects Both Fetus and Newborn. Neonatology 2013, 103, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Schindlbeck, U.; Höppner, S.; Kinting, S.; Frixel, S.; Kröner, C.; Liebisch, G.; Hegermann, J.; Aslanidis, C.; Brasch, F.; et al. Tools to explore ABCA3 mutations causing interstitial lung disease. Pediatr. Pulmonol. 2016, 51, 1284–1294. [Google Scholar] [CrossRef]
- Wambach, J.A.; Casey, A.M.; Fishman, M.P.; Wegner, D.J.; Wert, S.E.; Cole, F.S.; Hamvas, A.; Nogee, L.M. Genotype–Phenotype Correlations for Infants and Children with ABCA3 Deficiency. Am. J. Respir. Crit. Care Med. 2014, 189, 1538–1543. [Google Scholar] [CrossRef]
- Chowdhury, N.; Giles, B.L.; Dell, S.D. Full-Term Neonatal Respiratory Distress and Chronic Lung Disease. Pediatr. Ann. 2019, 48, e175–e181. [Google Scholar] [CrossRef]
- Bush, A.; Cunningham, S.; de Blic, J.; Barbato, A.; Clement, A.; Epaud, R.; Hengst, M.; Kiper, N.; Nicholson, A.G.; Wetzke, M.; et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax 2015, 70, 1078–1084. [Google Scholar] [CrossRef]
- Flamein, F.; Riffault, L.; Muselet-Charlier, C.; Pernelle, J.; Feldmann, D.; Jonard, L.; Durand-Schneider, A.-M.; Coulomb, A.; Maurice, M.; Nogee, L.M.; et al. Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung dis-eases in children. Hum. Mol. Genet. 2012, 21, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Wegner, D.J.; DePass, K.; Heins, H.; Druley, T.E.; Mitra, R.D.; An, P.; Zhang, Q.; Nogee, L.M.; Cole, F.S.; et al. Single ABCA3 Mutations Increase Risk for Neonatal Respiratory Distress Syndrome. Pediatrics 2012, 130, e1575–e1582. [Google Scholar] [CrossRef] [PubMed]
- Cheong, N.; Zhang, H.; Madesh, M.; Zhao, M.; Yu, K.; Dodia, C.; Fisher, A.B.; Savani, R.C.; Shuman, H. ABCA3 Is Critical for Lamellar Body Biogenesis in Vivo. J. Biol. Chem. 2007, 282, 23811–23817. [Google Scholar] [CrossRef] [PubMed]
- Garmany, T.H.; Moxley, M.A.; White, F.V.; Dean, M.; Hull, W.M.; Whitsett, J.A.; Nogee, L.M.; Hamvas, A. Surfactant Composition and Function in Patients with ABCA3 Mutations. Pediatr. Res. 2006, 59, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Yang, P.; Wegner, D.J.; Heins, H.B.; Luke, C.J.; Li, F.; White, F.V.; Silverman, G.A.; FCole, S.; Wambach, J.A. Functional Characterization of 4 ATP-Binding Cassette Transporter A3 Gene (ABCA3) Variants. Hum. Mutat. 2020, 41, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.-P.; Pinheiro, L.; Costa, M.; Silva, A.; Gonçalves, A.; Pereira, A. Novel ABCA3 mutations as a cause of respiratory distress in a term newborn. Gene 2014, 534, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Härtel, C.; Felderhoff-Müser, U.; Gebauer, C.; Hoehn, T.; Kribs, A.; Laux, R.; Möller, J.; Segerer, H.; Teig, N.; Von Der Wense, A.; et al. ATP-binding cassette member A3 (E292V) gene mutation and pulmonary morbidity in very-low-birth-weight infants. Acta Paediatr. 2011, 101, 380–383. [Google Scholar] [CrossRef]
- Garmany, T.H.; Wambach, J.A.; Heins, H.B.; Watkins-Torry, J.M.; Wegner, D.J.; Bennet, K.; An, P.; Land, G.; Saugstad, O.D.; Henderson, H.; et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr. Res. 2008, 63, 645–649. [Google Scholar] [CrossRef]
- Rezaei, F.; Shafiei, M.; Shariati, G.; Dehdashtian, A.; Mohebbi, M.; Galehdari, H. Novel Mutation in the ATP-Binding Cassette Transporter A3 (ABCA3) Encoding Gene Causes Respiratory Distress Syndrome in A Term Newborn in Southwest Iran. Iran. J. Pediatr. 2016, 26, 2493. [Google Scholar] [CrossRef]
- Wang, J.; Fan, J.; Zhang, Y.; Huang, L.; Shi, Y. ABCA3 gene mutations shape the clinical profiles of severe unexplained respiratory distress syndrome in late preterm and term infants. Transl. Pediatr. 2021, 10, 350–358. [Google Scholar] [CrossRef]
- Doan, M.L.; Guillerman, R.P.; Dishop, M.K.; Nogee, L.M.; Langston, C.; Mallory, G.B.; Sockrider, M.M.; Fan, L.L. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008, 63, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Szczapa, T.; Beck, J.; Migdal, M.; Gadzinowski, J. Monitoring diaphragm electrical activity and the detection of congenital central hypoventilation syndrome in a newborn. J. Perinatol. 2013, 33, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Ciantelli, M.; Ghirri, P.; Presi, S.; Sigali, E.; Vuerich, M.; Somaschini, M.; Ferrari, M.; Boldrini, A.; Carrera, P. Fatal respiratory failure in a full-term newborn with two ABCA3 gene mutations: A case report. J. Perinatol. 2010, 31, 70–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nishida, D.; Kawabe, S.; Iwata, N.; Cho, K. ABCA3 deficiency dramatically improved by azithromycin administration. Pediatr. Int. 2021, 63, 602–604. [Google Scholar] [CrossRef]
- Kitazawa, H.; Moriya, K.; Niizuma, H.; Kawano, K.; Saito-Nanjo, Y.; Uchiyama, T.; Rikiishi, T.; Sasahara, Y.; Sakamoto, O.; Setoguchi, Y.; et al. Interstitial lung disease in two brothers with novel compound heterozygous ABCA3 mutations. Eur. J. Pediatr. 2013, 172, 953–957. [Google Scholar] [CrossRef]
- Williamson, M.; Wallis, C. Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr. Pulmonol. 2014, 49, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Kröner, C.; Wittmann, T.; Reu, S.; Teusch, V.; Klemme, M.; Rauch, D.; Hengst, M.; Kappler, M.; Cobanoglu, N.; Sismanlar, T.; et al. Lung disease caused by ABCA3 mutations. Thorax 2017, 72, 213–220. [Google Scholar] [CrossRef]
- Si, X.; Steffes, L.C.; Schymick, J.C.; Hazard, F.K.; Tracy, M.C.; Cornfield, D.N. Three Infants with Pathogenic Variants in the ABCA3 Gene: Presentation, Treatment, and Clinical Course. J. Pediatr. 2021, 231, 278–283. [Google Scholar] [CrossRef]
- López MD, C.; Ruiz, E.P.; Galdo, A.M.; Torrent-Vernetta, A.; Deu, M.; Ramirez, J.B.; Vives, M.A.R.; Aguilera, P.C.; Frías, F.J.P. ABCA3 Deficiency in a Newborn with Respiratory Failure. Arch. Bronconeumol. 2018, 54, 634–635. [Google Scholar]
- Eldridge, W.B.; Zhang, Q.; Faro, A.; Sweet, S.C.; Eghtesady, P.; Hamvas, A.; Co, F.S.; Wambac, J.A. Outcomes of Lung Transplantation for Infants and Children with Genetic Disorders of Surfactant Metabolism. J. Pediatr. 2017, 184, 157–164. [Google Scholar] [CrossRef]
- De Luca, D.; Tissieres, P.; Kneyber, M.C.J.; Humbert, M.; Mercier, O. Lung transplantation in neonates and infants: ESPNIC survey of European neonatologists and pediatric intensivists. Eur. J. Pediatr. 2021, 180, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Griese, M. Chronic interstitial lung disease in children. Eur. Respir. Rev. 2018, 27, 170100. [Google Scholar] [CrossRef] [PubMed]
- Forstner, M.; Lin, S.; Yang, X.; Kinting, S.; Rothenaigner, I.; Schorpp, K.; Li, Y.; Hadian, K.; Griese, M. High-Content Screening Identifies Cyclosporin A as a Novel ABCA3-Specific Molecular Corrector. Am. J. Respir. Cell Mol. Biol. 2021, 66, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Nogee, L.M.; Cole, F.S. First Steps Toward Personalized Therapies for ABCA3 Deficiency. Am. J. Respir. Cell Mol. Biol. 2022, 66, 349–350. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).