
Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction and Goals

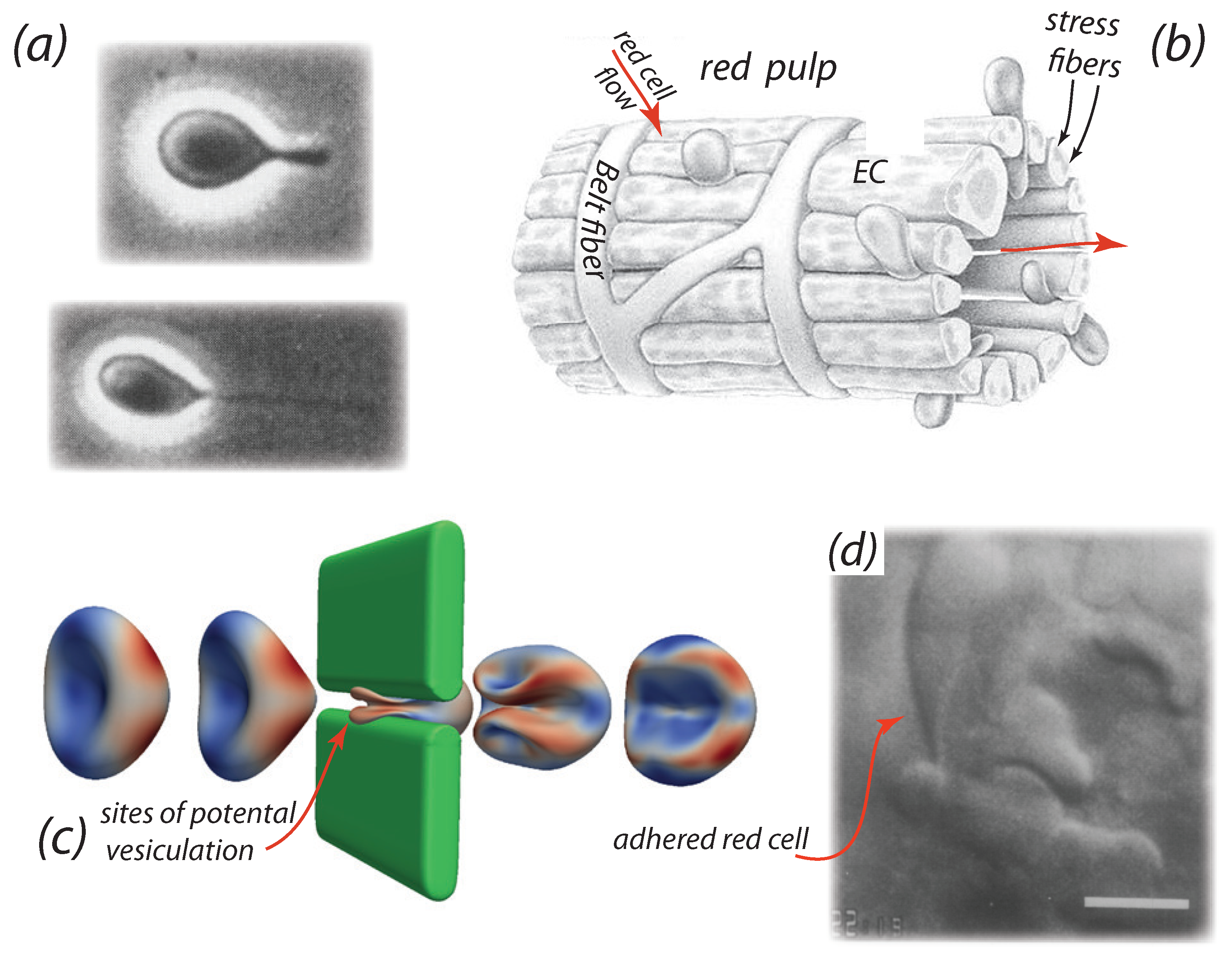
2. Background on the Splenic Example
- The video microscopy of MacDonald et al. [89,99] shows that venous slits open in “bursts” of durations of ∼10 s; moreover adjacent slits open and close in an asynchronous manner. Mediators of this behavior may include contraction of stress fibers [95,96,97] that do so in an unsymmetrical fashion [74]
- Additional cell passage through a given slit, that has trapped a cell “by its tail”, indeed occurs which demonstrates that slits are not simply “propped open” by a passing cell. See Figure 1d for an example. Time scales of cell trapping events were recorded by MacDonald et al. [89] to be of at least and were observed to persist for as long as 10 s in some cases.
- Cells passing through a slit with a trapped cell in place do not, or may not, adhere to the previous and trapped cell. Note that PS exposure on the outer membrane of erythrocytes is known to promote RBC adhesion [14,15,28,106,107] and hence the occurrence of RBC adhesion may be quite RBC specific - i.e., some adhere and some do not, since some cells expose PS and some do not.
- Trapping of cells by adhesion may then be related to the exposure of PS on its outer membrane leaflet, a feature known to promote adhesion to vascular endothelial cells [28,106,107,108] or by activation of Lu/BCAM-laminin-α5 adhesion [41,42]. PS exposure, which involves the loss of lipid concentration asymmetry, can provide a direct driver of vesiculation [44]. In addition, we note that Willekens et al. [109] have hypothesized that haemoglobin loss from erythrocytes can result from spleen facilitated vesiculation, perhaps we add in the manner recently proposed by Klei et al. [41].
- Still again, the electronegativity of glycocalyx of these cells [110] may also induce adhesion. This is discussed below in brief as pertains, for example, to the effects of partial cancelation of repulsive negative charges on the glycocalyx of red and endothelial cells that, in turn, normally imparts a low friction between these cells and thereby allows for the free flow of RBCs.
3. Hypothesis: Vesiculation Due to RBC-EC Interaction
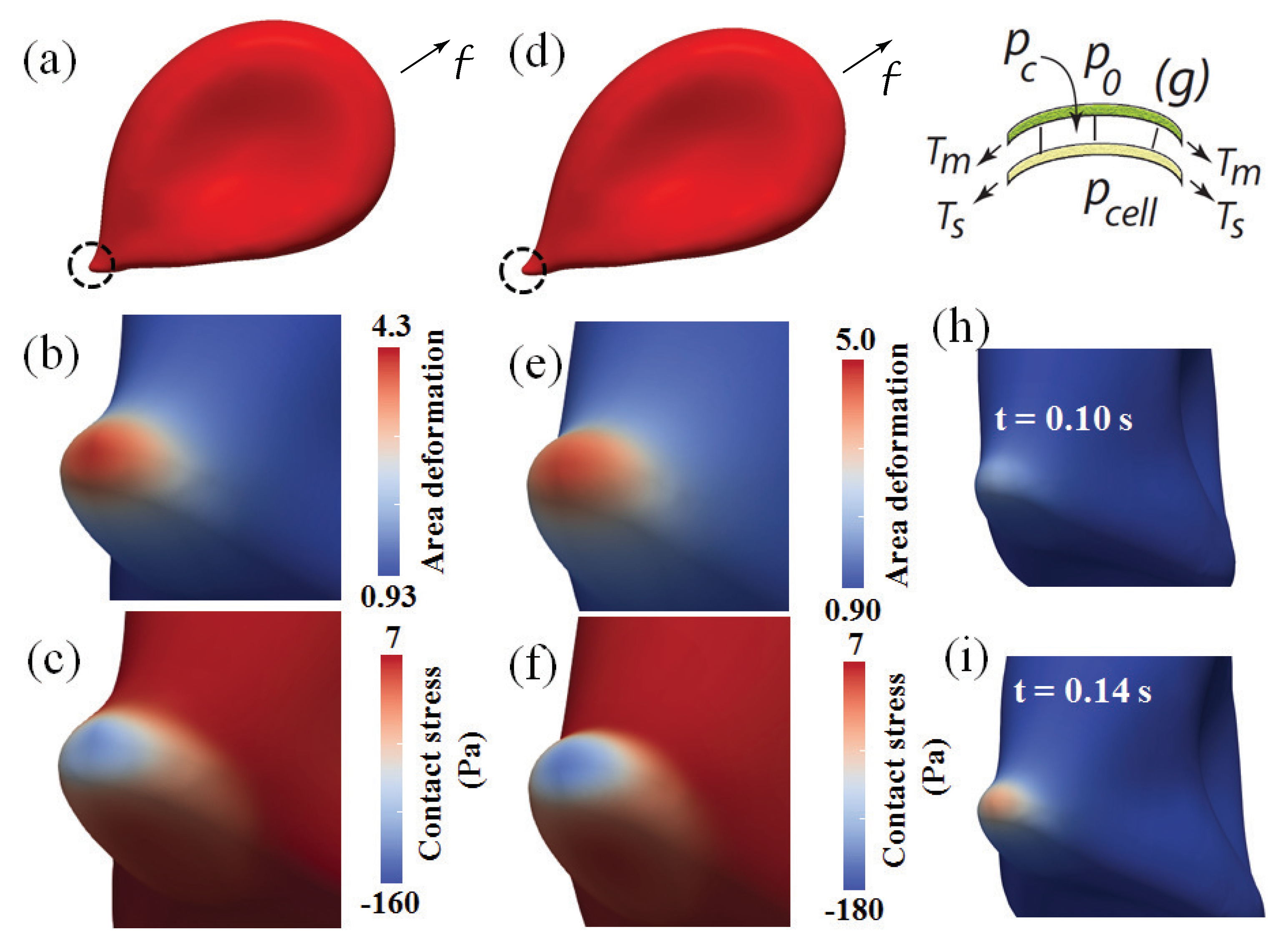
3.1. Simulation Results: Cell Adhesion and Tethering
3.1.1. Overall Cell Deformation and Forces
3.1.2. Skeletal Remodeling via Area Deformation: Time Scaling
3.1.3. Contact Pressure and Stress
3.1.4. Summary Assessment of Vesiculation Prospects
4. RBC Adherence to Endothelial Cells: Pathology and Aging
- Kucukal et al. [121,122] and Kaul [123] have reviewed the role played by red blood cell adhesion in sickle cell disease while Jambou et al. [124] have described a trogocytosis-like mechanism for the interaction of malaria infected red blood cells with human brain endothelial cells. Moreover, Wautier and Wautier [108] and Zwaal et al. [125] have reviewed research dealing focused on, and the molecular basis for, increased erythrocyte adherence to vascular endothelial cells; this included research that demonstrated enhanced adherence due to PS exposure that provided a molecular basis for enhanced RBC adherence in pathologies such as diabetes mellitus and central retinal vein occlusion [28]. Although their measurements of “adhesive strength” are of a qualitative sort, and hence precise stress levels that the adherent cells might bare unknown, it appeared that sustainable traction stress levels in excess of (10–20 Pa) were clearly in the range [126]. Three examples are briefly discussed, followed by an additional example of a quite different type. Kucukal et al. [127], however, have preformed microfluidic testing on adhered sickle cell disease cells and using shear rates that well exceeded s−1; this would place the shear stress we quote as a factor of 5 above the range we demonstrated vesiculation to be forecasted at.
- (a)
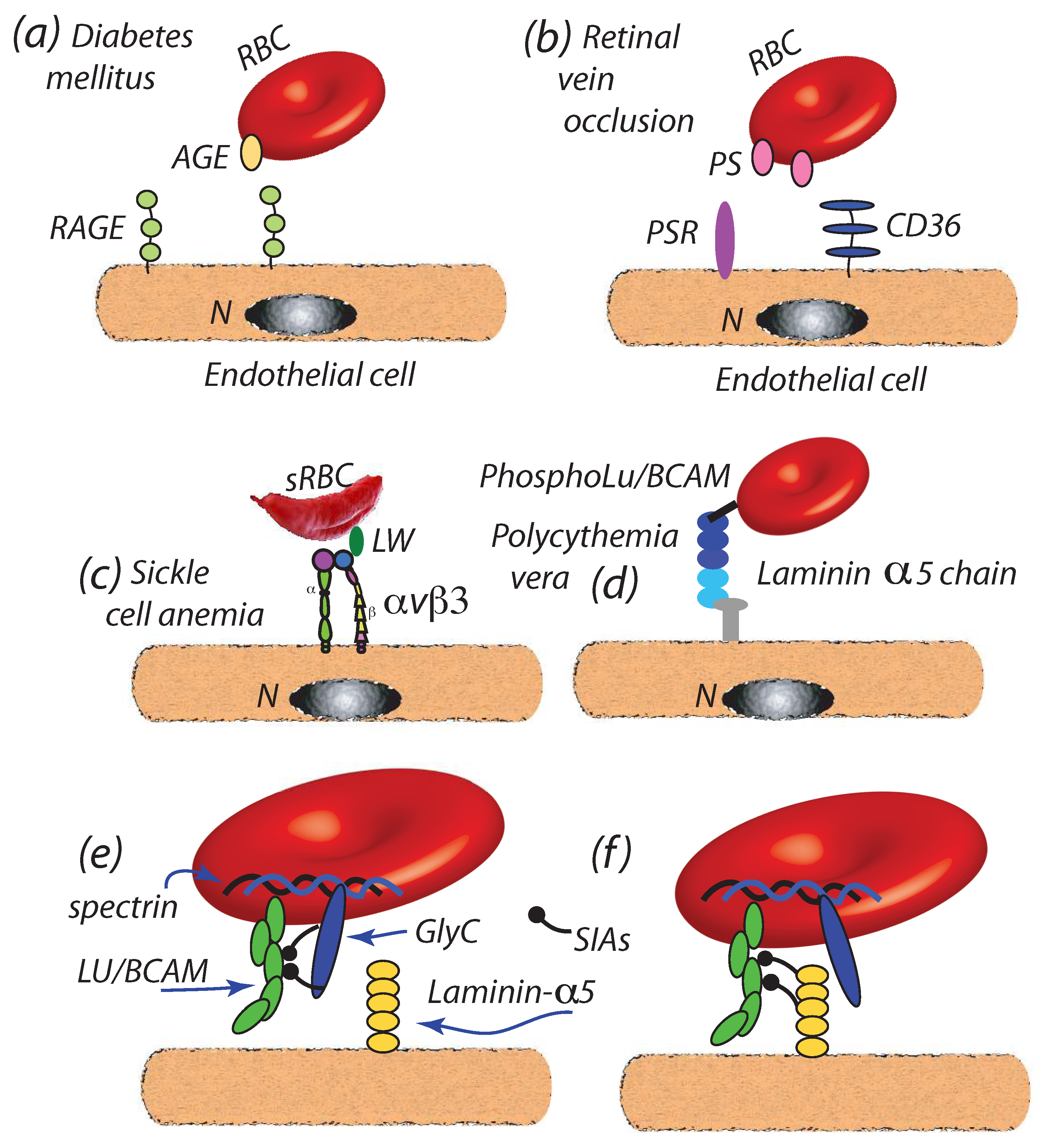
- Diabetes mellitus: Wautier et al. [36] have reported that enhanced cell-endothelial adherence evolved in parallel with glycated hemoglobin HbA1c, wherein we note that HbA1c content is indeed found to increase in RBCs during the normal, i.e., healthy, aging process [128]; HbA1c levels have been, in fact, recommended as a diagnostic marker for diabetes mellitus [129]. The resultant Advanced Glycation End products (AGEs) so produced are ligated to the receptor for AGEs (RAGE) of endothelial cells [37,130,131,132] as depicted in Figure 4a. Full characterization of such binding is, as yet, outstanding.We note that increased levels of HbA1c have been positively correlated with plasma VEGF (Vascular Endothelial Growth Factor) levels and hence, that is via the influence of VGEF in angiogenesis [133]. Such effect may contribute to the progression of atherosclerosis in diabetic patients [11,134]. It is also reported that vesicle concentrations are increased with type 2 diabetes, where the role of vesicles containing inflammatory cargo, e.g., HbA1c, has also been highlighted in these recent studies [9,10,11]. Although the mechanistic cause for the VEGF-HbA1c correlation is as yet unclear, it may be suspected that HbA1c laden RBC-derived vesciles may be the trigger for unregulated VEGF expression. The general schema of such a mechanism may be as described by the red cell cross-talk depicted in Figure 5 which is discussed below, specifically in connection with atherosclerosis.
- (b)
- Retinal vein occlusion (RVO): retinal vein occlusion (RVO) is a common cause of permanent vision loss. Wautier and co-workers [28] reported that PS exposure was enhanced in RVO patients and correlated with enhanced cell-endothelium adhesion; the mechanism postulated was binding to PS receptors (PSR) of endothelial cells as depicted in Figure 4b; see also [27,107,135] for additional discussion of PS enhanced adhesion. When vascular leakage occurs, due to increased intraluminal pressure, this may lead to ischemia and the secretion of VEGF that may cause further leakage and retinal edema [29,136]. Hence, VEGF appears to play a significant role in RVO progression. It may be noted that enhanced PS exposure is also commonly found in other pathologies such as sickle cell anemia [123,137] wherein enhanced cell adhesion is also observed.
- (c)
- Sickle cell disease (SCD): recent studies show that the critical flow shear stress to detach a sickle mature erythrocyte in oxygenated is 3.9–5.5 Pa in oxygenated state and above 6.2 Pa in deoxygenated state, although multiple adhesion sites may be involved [32]. In SCD a number of proteins have been implicated in adhesion to the endothelium including integrins such as or binding to receptors such as adhesion molecule-4 (LW) and CD36 [33,34,35,138,139], as depicted in Figure 4c. It is known that adhesion in such cases can support traction stresses well in excess of 10–100 Pa and larger [126] and hence readily support binding as envisioned herein. In fact, Kaul et al. [33] found that blocking “may constitute a potential therapeutic approach to prevent SS RBC-endothelium interactions under flow conditions”.
- (d)
- Polycythemia vera: Polycythemia vera (PV) is a chronic disorder characterized by an increase in red cell mass resulting in hyperviscosity [19,20]. PV is also characterized by an increase in adhesiveness of red cells to the endothelium [19]; the adhesion molecule Lu/BCAM is a main player in the adhesion as depicted in Figure 4d [19,21]. It is of interest to note that Wautier et al. [19] have observed that PV cells remained adhered to human umbilical vein endothelial cells within the shear stress range Pa Pa thus demonstrating a resistance beyond the critical range of Pa found by Hochmuth et al. [72] at which tethers and evagination formed in healthy erythrocytes and the Pa that Franco et al. [26] found induced tethering in Gaucher disease RBCs (see below). Still another unaddressed line of inquiry is that, since phosphorylation of the cytoplasmic domain of Lu/BCAM is required to activate its extracellular domain, the interaction of Lu/BCAM with the erythrocyte skeleton may be quite relevant to the integrity of the M⇆S connectivity.Increased concentrations of microvesicles are indeed reported with PV [12,22,140] and their role in inducing thrombosis in PV noted. Tan et al. [12] specifically discuss the correlation of exposed PS on PV erythrocytes and platelets and point to the correlations of Morel et al. [141] of PS exposure and vesiculation of RBCs; biophysical mechanisms for this effect of PS have been detailed by Asaro et al. [44]. Kroll et al. [22] further point to the association of JAK2 with unregulated hematopoiatic proliferation and thrombotic risk; this is discussed further in connection with the link between erythrocute-derived microvesicles and myeloproliferative neoplasm [31].
- (e)
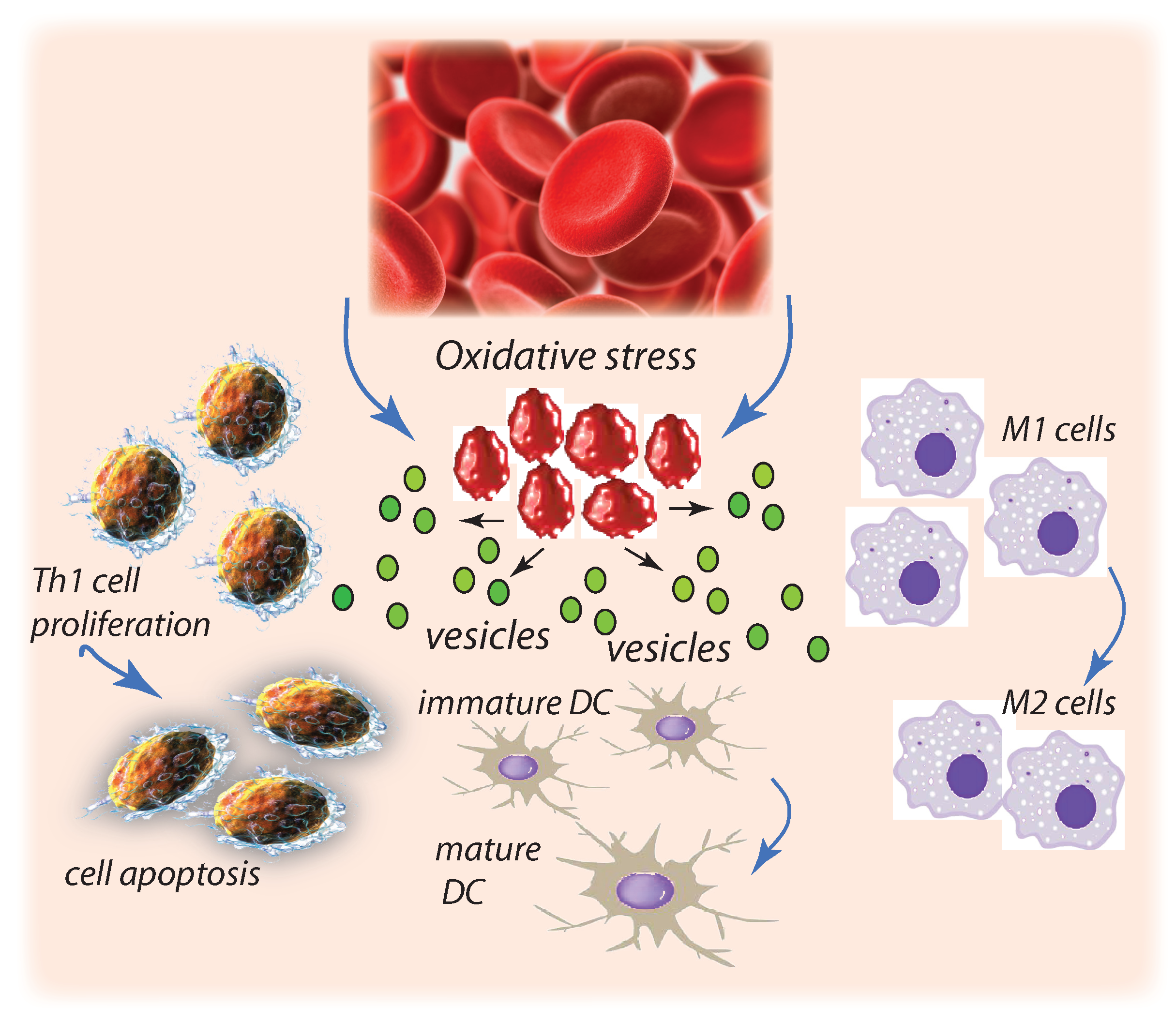
- Myeloproliferative neoplasms: Myeloproliferative neoplasms (MPNs) are hematopoietic stem cell dysfunctions that lead to over proliferation of particular cell linages. These include, inter alia, myelogenous leukemia (granulocytes); PV (RBCs); essential thrombocythemia (platelets); and neutrophilic leukemia (white cells) [142]. This extensive topic is beyond the scope of the present focused review but we note that recent findings of Poisson et al. [31] who demonstrated that vesicles derived from JAK2 erythrocytes induce ROS and vascular endothelial constriction. In fact, Poisson et al.’s [31] Abstract illustration portrays a schema of the role of microvesicles quite reminiscent of Figure 1 of Buttari et al. [1] that we have adapted for our Figure 5. It remains to be shown if, aside from the increase in RBCs in PV, that the prospects for vesiculation in MPN RBCs is also increased. The links between those factors that lead to increased RBC adhesion and decreased membrane-skeleton connectivity and our vesiculation paradigm suggest this may well be the case.
- (f)
- Gaucher disease: Gaucher disease (GD) which is caused by glucocerbrosidase deficiency and is also characterized by increased RBC adherence to endothelial cells, also involves LuBCAM [26]. The study by Franco et al. [26] is particularly notable here since in their study of GD red cells, they reported that when shear flow shear stresses were raised to levels at or above Pa GD cells “exhibited frequent and elongated membrane tethers” (see their Figures 5b,c in [26]). Results for shear stresses below that range were not reported except to note that at Pa cells were reported to take on only racket-like shapes (see their Figure 5a); these were, in fact, quite similar to the simulated cell shapes of Figure 3a,d. They hypothesized that this suggested that the skeletal-membrane connectivity was altered and favored “membrane dissociation from the skeleton” [26]. Hence while adhered in a shear flow field their GD RBCs conformed to our proposed paradigm and these observations, indeed, warrant more systematic study; it is not known what the critical shear stress levels for tether formation were for GD vs. healthy red cells. Bratosin et al. [30] report that GD red blood cells do expose PS and reduced CD47, a self-recognition marker protecting against phagocytosis; this along with their abnormal shapes may mark them for early clearance, in particular in the spleen.
- It is understood that the electronegativity of the glycocalyx of erythrocytes and endothelial cells facilitate low friction blood flow [143,144,145,146]. Accordingly, Oberieithner et al. [110] performed a series of AFM probes involving de-cohering RBCs for cultured endothelial cells in the presence of elevated , intended to reduce or neutralize net negative charge. In this they recorded increased adherence at sufficiently high concentrations. Although of a exceedingly qualitative nature, as contact areas were not measured, nor was the geometry of “peel” profiles, they reported maximum extraction forces of pN). Hence, even considering contact areas as large as 50–100 , nominal stresses would have been greater than the range 10–50 Pa.We add still another comment concerning the possible role of the RBC glycocalyx regarding cell adherence, in this case cell aging. Neu et al., [147] have argued, with supporting analysis, that changes in the erythrocyte glycocalyx, in particular in its thickness, may alter the electrostatic fields between cells that affects their affinity with respect to aggregation. This view overlaps that noted above regarding the reduction of surface electronegativity that, in turn, reduces repulsion between cells and enhances adhesion. They further point out that for aged cells, which are targets for macrophage removal, that that process itself requires macrophage recognition and association, a process that requires close proximity of these cells; to accomplish close proximity macrophages and erythrocytes must overcome repulsion of a thick glycocalyx [148]. The electrostatic model they use is, in fact, quite similar to that developed by Zhu et al. [98] to describe resistance of an erythrocyte glycocalyx to compression as it attempts to squeeze through a venous slit. Hence we note the possible connections between RBC adhesion and senescence to pathologies and aging per se that affect the RBC glycocalyx, a topic that requires additional inquiry as Neu et al. indeed point out [147].
- Atherosclerosis: red cell cross talk: atherosclerosis, a chronic progressive, multifactorial disease may be another that involves the process of erythrocyte adhesion to the endothelium within blood vessels [4,149,150,151]. The manner in which red cells may help mediate the formation and destabilization of plaque is via crosstalk between innate and adaptive immune cells, macrophages, and T lymphocytes [1,149]. For example, immune cells may be activated by various endogenous molecules that have undergone chemical or structural changes; this may include haemoglobin released from lysed RBCs or from RBC-derived vesicles [1]. Moreover, exosomes released from RBCs bind to monocytes and induce proinflammatory cytokines that boost T-cell response [150]. Red cell activity would be enhanced by oxidative stress that occurs during aging and is known to be prevalent during red cell storage [47,152]. Oxidatively stressed red cells expose PS and contain aggregated and/or oxidized HB which is released during the process of vesiculation [47]. The mechanisms we propose may indeed contribute to this process and possible mechanisms of red cell crosstalk are depicted in Figure 5 as adapted from Buttari et al. [1].For example, oxidized RBCs that do not control lipolysaccharide (LPS)-induced dentritic cell (DC) maturation promote DC maturation to a proinflammatory TH1 cell response [151,153]. A mechanism for this is the loss of CD47 at the erythrocyte surface due to vesiculation; CD47 appears to be critical to the role RBCs play preventing DC maturation [153]. Stored RBCs may polarize macrophages toward the M1 pathway associated with proinflammatory cytokine production. Oxidative damage promotes disruption of RBC membrane-skeleton connectivity that, in turn, induces vesiculation [44]. In addition, extracellular vesicles released by RBCs, e.g., during storage, appear capable of stimulating Th1 cells and provoking proinflammatory response by releasing cytokines [3,154].The role of erythrocyte-derived vessels and release of Hb in atherosclerotic lesions, and in particular within plaque neovessels [8], has been discussed with pointed reference to their effects on coagulation, inflammatory response, as well as cell adhesion per se [6,7,16,17,150]. Red cells may vesiculate more readily within the oxidative environment of atherosclerotic plaques due, in part, to the hypoxia that exists; because of hypoxic conditions there may be a switch from aerobic to anaerobic metabolism, characterized by glucose and ATP depletion [68,155], conditions that are known to promote erythrocyte vesiculation [44,64,152,156,157,158,159]. Erythrocyte-derived vesicles can then release Hb and contribute to the generation of such species as metHb and oxHb. This again suggests that vesicles produced during microvascular flow local to atherosclerotic plaques may indeed participate in plaque progression and destabilization.In addition, Jenny et al. [8] and Tziakas and co-workers [160,161] have hypothesized that cholesterol as well as Hb released by RBCs within atherosclerotic plaques are important contributors to plaque instability, wherein the latter refer to a mechanism involving the “breakdown of erythrocytes” releasing cholesterol; the release of Hb containing vesciles may well be part of this process although the number of vesicles would be most likely quite large to account for the cholesterol levels estimated. The possible, and probable, roles of RBC-derived vesicles in destabilizing plaques have been further reviewed by Boulanger et al. [17]. They point out the general trends of increased microvesicle levels with a range of cardiovascular risk factors including, inter alia, dyslipaemia, diabetes mellitus, hypertension, and atherosclerosis. The role of platelet derived vesicles in the development of atherosclerosis in diabetes mellitus has been discussed, in fact, for over 25 years [159].Taken together, the above suggests that oxidatively stressed RBCs that contain oxidized Hb, externally expose PS, and have compromised membrane-skeleton connectivity, may undergo increased endothelial cell adhesion and vesiculation via the mechanisms discussed herein. Such vesicles may then engage in cross-talk as discussed by Buttari et al. [1] thereby impacting the progression of atherosclerosis.
5. Discussion
5.1. Splenic Red Blood Cell Clearance
5.2. Studies of Cell-Cell Adhesive Interaction under Shear Flow
- Use of microfluidic shear flow chambers as a general set up: the studies of Hochmuth et al. [72] have already been cited; their methods are simple yet provide a useful methodology to explore the mechanisms of tether formation and possible vesiculation. It would be advisable to extend such methods to follow the pathways of hemolysis. Additional methods include those of Yang et al. [174] who studied red cell adhesion to endothelial cells via a microfluidics approach and by Alapan et al. [175] or Kucukal et al. [127] who studied adhesive effects in sickle cell disease RBCs. The latter methods provide for greater throughput and enhanced visualization. Still other microfluidic devices offer either single cell or high throughput measurement within highly controlled environments [127,176,177,178,179] and should be considered.
- Vesiculation and lysis of senescent RBCs: the hypothesis that senescent red cells undergo hemolysis, while adhered to the red pulp, via a loss in sialic acid and under shear flow would benefit from additional mechanistic understanding. For example, questions such as, “where in the red pulp does hemolysis occur?” and “how does the bleeding of hemoglobin occur?” are relevant, or even “do membrane rafts paly a role in vesiculation leading to hemolysis [180]?” Figure 1d shows a red cell trapped as it passes through an IES; this is precisely the process analyzed by Asaro et al. [74] as reviewed by Figure 3. However, once a red cell passes through an IES into the sinus lumen, it is unclear how lysis would then occur thereafter. Hence it may be important to understand how hemolysis occurs under more modest shear flow as red cells approach, and are trapped for various times yet unknown, in more modest shear flows in the splenic red pulp. The theoretical perspective of the analysis and simulations of Asaro et al. [74] are quite relevant here as well as they provide quantitative background on details such as membrane tension and skeletal remodeling that are important for assessing pore development involved in hemoglobin loss.This suggests that studies involving red cells that are treated to be senescent-like as described by Klei et al. [41], and others [181,182,183,184], and that are depleted in sialic acid to induce LU/BCAM-laminin- adhesion, would be advisable. The microfluidic test set-ups described above may provide for an adequate methodology.
5.3. Implications for Clinical Diagnosis
6. Compliance with Ethical Standards
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Buttari, B.; Profumo, E.; Riganò, R. Crosstalk between Red Blood Cells and the Immune System and Its Impact on Atherosclerosis. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Loyer, X.; Rautou, P.-E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef]
- Kim-Shapiro, D.B.; Lee, J.; Gladwin, M.T. Storage lesion: Role of red blood cell breakdown. Transfusion 2011, 51, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Businaro, R.; Tagliani, A.; Buttari, B.; Profumo, E.; Ippoliti, F.; Cristofano, C.D.; Capoano, R.; Salvati, B.; Riganò, R. Cellular and molecular players in the atherosclerosis plaque progression. Ann. N. Y. Acad. Sci. 2012, 1262, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Jy, W.; Ricci, M.; Shariatmadar, S.; Gomez-Marin, O.; Horstman, L.H.; Ahn, Y.S. Microparticles in stored red blood cells as potential mediators of transfusion complications. Transfusion 2011, 51, 886–893. [Google Scholar] [CrossRef]
- Li, K.Y.; Zheng, L.; Qang, Q.; Hu, Y.W. Characteristics of erythrocyte-derived microvessels and its relation with atherosclerosis. Atherosclerosis 2016, 255, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Guérin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C. Circulating endothelial microparticles are associated with vascular dysfuction in patients with end-stage renal failure. Am. Soc. Nephrol. 2005, 1046, 3381–3388. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.W.; Hooten, N.N.; Eitan, E.; Green, J.; Mode, N.A.; Bodogai, M.; Zhang, Y.; Lehrmann, E.; Zonderman, A.B.; Biragyn, A.; et al. Altered Extracellular Vesicle Concentration, Cargo, and Function in Diabetes. Diabetes 2018, 67, 2377–2388. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, L.; Zou, X.; Wang, J.; Zhong, J.; Zhong, T. Extracellular vesicles in type 2 diabetes mellitus: Key roles in pathogenesis, complications, and therapy. J. Extracell. Vesicles 2019, 8, 1625677. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Hooten, N.C.; Freeman, D.W.; Mode, N.A.; Zonderman, A.B. Extracellular vesicles in diabetes mettitus induce alterations in endothelial cell morphology and migration. J. Transl. Med. 2020, 18, 230–246. [Google Scholar] [CrossRef]
- Gao, C.; Yang, X.; Li, J.; Wang, W.; Hou, J.; Li, H.; Tan, X.; Shi, J.; Fu, Y.; Zhou, J. Role of erythrocytes and platelets in the hypercoagulable status in polycythemia vera through phosphatidylserine exposure and microparticle generation. Thromb. Haemost. 2013, 109, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Kriebardis, M.H.; Antonelou, K.E.; Stamoulis, E.; Economou-Petersen, E.; Margaritis, I.H.; Papassideri, I.S. RBC-derived vesicles during storage untrastructure, protein composition, oxidation, and singling components. Transfusion 2008, 48, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.L.; Boilard, E.; Buzas, E.I.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in vascular homeostatis and diseases. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar]
- Zarà, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Extracellular vesicles and atherosclerosis disease. Cell. Mol. Life Sci. 2015, 72, 2697–2708. [Google Scholar] [CrossRef] [PubMed]
- Loyer, V.; Vion, A.C.; Tedgui, A.; Boulanger, C.M. Microvessels as cell-cell messengers in cardiovascular diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P. Cellular and molecular aspects of blood cell-endothelium interactions in vasular disorders. Int. J. Mol. Sci. 2020, 21, 5315. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Nemer, W.E.; Gane, P.; Rain, J.D.; Carton, J.P.; Colin, Y.; Kim, C.L.V.; Wautier, J.L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythmia vera is mediated by laminin chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Nemer, E.; Grandis, W.M.D.; Brusson, M. Abnormal adhesion of red blood cells in polycythemia vera: A prothrombotic effect? Thromb. Res. 2014, 133, S107–S111. [Google Scholar] [CrossRef]
- De Grandis, M.; Cambot, M.; Wautier, M.-P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.-L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.H.; Michaelis, L.C.; Verstovsek, S. Mechanisms of thrombogenesis in polycythemia vera. Blood Rev. 2015, 29, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Krieglstein, C.F.; Granger, D.N. Adhesion molecules and their role in vascular disease. Am. J. Hypertens. 2001, 14, 44S–54S. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; Bevilacqua, M.P.; Cybulsky, M.I. Endothelial-Dependent Mechanisms of Leukocyte Adhesion in Inflammation and Atherosclerosis. Ann. N. Y. Acad. Sci. 1990, 598, 77–85. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Allen, M.D.; O McDonald, T.; Chait, A.; Harlan, J.M.; Fishbein, D.; Mccarty, J.; Ferguson, M.; Hudkins, K.; Benjamin, C.D. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J. Clin. Investig. 1993, 92, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Collec, E.; Connes, P.; Akker, E.V.D.; De Villemeur, T.B.; Belmatoug, N.; Von Lindern, M.; Ameziane, N.; Hermine, O.; Colin, Y.; et al. Abnormal properties of red blood cells suggest a role in the pathophysiology of Gaucher disease. Blood 2013, 121, 546–555. [Google Scholar] [CrossRef]
- Pretini, V.; Koenen, M.H.; Kaestner, L.; Fens, M.H.A.M.; Schiffelers, R.M.; Bartels, M.; Van Wijk, R. Red Blood Cells: Chasing Interactions. Front. Physiol. 2019, 10, 945. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Hé, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in certral retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.M.; Dugel, P.U.; Chen, S.; Blinder, K.J.; Walt, J.G. Anti-VEGF treatment of macular edema associated with retinal vein occlusion: Patterns of use and effectiveness in clinical practice (ECHO study report 2). Clin. Ophthalmol. 2018, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Bratosin, D.; Tissier, J.-P.; Lapillonne, H.; Hermine, O.; De Villemeur, T.B.; Cotoraci, C.; Montreuil, J.; Mignot, C. A cytometric study of the red blood cells in Gaucher disease reveals their abnormal shape that may be involved in increased erythrophagocytosis. Cytom. Part B Clin. Cytom. 2010, 80, 28–37. [Google Scholar] [CrossRef]
- Poisson, J.; Tanguy, M.; Davy, H.; Camara, F.; El Mdawar, M.-B.; Kheloufi, M.; Dagher, T.; Devue, C.; Lasselin, J.; Plessier, A.; et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Investig. 2020, 130, 2630–2643. [Google Scholar] [CrossRef]
- Deng, Y.; Papageorgiou, D.P.; Chang, H.-Y.; Abidi, S.Z.; Li, X.; Dao, M.; Karniadakis, G.E. Quantifying Shear-Induced Deformation and Detachment of Individual Adherent Sickle Red Blood Cells. Biophys. J. 2019, 116, 360–371. [Google Scholar] [CrossRef]
- Kaul, D.K.; Tsai, H.M.; Liu, X.D.; Nakada, M.T.; Nagel, R.L.; Coller, B.S. Monoclonal antibidies to a αvβ3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by platlet activating factor. Blood 2000, 95, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Zennadi, R.; Hines, P.C.; de Castro, I.M.; Cartron, J.P.; Parise, I.V.; Telen, M.J. Epinephrine acts through erythroid singling pathways to activate sickle cell adhesion to endothelium via LW-αvβ3 interactions. Blood 2004, 104, 3774–3781. [Google Scholar] [CrossRef] [PubMed]
- Telen, M. Role of adhesion molecules and vascular endothelium in the pathogenesis of sickle vell disease. Hematol. Am. Soc. Educ. Program 2007, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Leblanc, H.; Wautier, M.P.; Abadie, E.; Passa, P.; Caen, J.P. Erythrocyte adhesion to cultured endothelium and glycaemic control in Type I (insulin-dependent) diabetic patients. Diabetologia 1986, 29, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Grossin, N.; Wautier, M.-P.S.; Picot, J.; Stern, D.; Wautier, J.-L.T. Differential effect of plasma or erythrocyte AGE-ligands of RAGE on expression of transcripts for receptor isoforms. Diabetes Metab. 2009, 35, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Willekens, F.L.A.J.; Weere, M.; Groenen-Döpp, Y.A.; Bregt-Roerdinkholder, B.; de Pauw, B.; Bosman, G.J.C.G.M. Erythrocyte vesiculation: A self-protective mechanism? Br. J. Haematol. 2003, 141, 549–556. [Google Scholar] [CrossRef]
- Ciana, A.; Achilli, C.; Gaur, A.; Minetti, G. Membrane Remodelling and Vesicle Formation During Ageing of Human Red Blood Cells. Cell. Physiol. Biochem. 2017, 42, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Ciana, A.; Achilli, C.; Minetti, G. Spectrin and Other Membrane-Skeletal Components in Human Red Blood Cells of Different Age. Cell. Physiol. Biochem. 2017, 42, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; Dalomot, J.; Nota, B.; Veldthuis, M.; Rademakers, F.P.J.M.T.; Hoogenboezem, M.; Nagelkerke, S.Q.; Jcken, W.F.J.V.; Oole, E.; Svendsen, P.; et al. Hemolysis in the spleen drives erythrocyte turnover. Blood 2020, 136, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; de Back, D.Z.; Asif, P.J.; Verkijlen, P.J.J.H.; Veldthuis, M.; Lighart, P.C.; Berghuis, J.; Clifford, E.; Beuger, B.M.; Berg, T.K.V.; et al. Glycophorin-C sialyation regulates Lu/BCAM adhesive capacity during eruthrocyte aging. Blood Adv. 2018, 2, 14–24. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; McRae, K.; Lam, F.; McAvoy, E.F.; Clark, S.; Brain, M.; Lalor, P.F.; Adams, D.H.; Kubes, P. A novel mechanism of erythrocyte capture from circulation in humans. Exp. Hematol. 2008, 36, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Asaro, R.J.; Zhu, Q.; Cabrales, P. Erythrocyte Aging, Protection via Vesiculation: An Analysis Methodology via Oscillatory Flow. Front. Physiol. 2018, 9, 1607. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Kawashima, H.; Tanaka, M. Analysis of erythrocyte membrane proteins in patients with hereditary spherocytosis and other types of haemolytic anaemia. Hematology 2018, 23, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Alaarg, A.; Schiffelers, R.M.; van Solinge, W.W.; van Wijk, R. Red blood cell vesiculation in hereditary hemolytic anemia. Front. Physiol. 2013, 4, 365. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.K.; Adjobo-Hermans, M.J.W.; Bosman, G.J.C.M. Red blood cell Homeostatis: Mechanics and effects of microvesicle generation in health and disease. Front. Physiol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G. Red Cell Membrane Disorders. Hematology 2005, 2005, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G. Update on the clinical spectrum andgenetics of red blood cell membrane disorders. Curr. Hematol. Rep. 2004, 3, 85–91. [Google Scholar]
- Walensky, L. Disorders of the Red Blood Cell Membrane; Lippincott, Williams and Williams: Philadelphia, PA, USA, 2003; pp. 1709–1858. [Google Scholar]
- An, X.; Mohandas, N. Disorders of red cell membrane. Br. J. Haematol. 2008, 141, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Spangler, E.J.; Harvey, C.W.; Revalee, J.D.; Kumar, P.B.S.; Laradji, M. Computer simulation of cytoskeleton-induced blebbing in lipid membranes. Phys. Rev. E 2011, 84, 051906. [Google Scholar] [CrossRef]
- Li, H.; Yang, J.; Chu, T.T.; Naidu, R.; Lu, L.; Chandramohanadas, R.; Dao, M.; Karniadakis, G.E. Cytoskeleton Remodeling Induces Membrane Stiffness and Stability Changes of Maturing Reticulocytes. Biophys. J. 2018, 114, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lykotrafitis, G. Vesiculation of healthy and defective red blood cells. Phys. Rev. E 2015, 92, 012715. [Google Scholar] [CrossRef] [PubMed]
- Parkar, N.S.; Akpa, B.S.; Nitsche, L.C.; Wedgewood, L.E.; Place, A.T.; Sverdlov, M.S.; Chaga, O.; Minshall, R.D. Vesicle Formation and Endocytosis: Function, Machinery, Mechanisms, and Modeling. Antioxid. Redox Signal. 2009, 11, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S. Clathrin-Coated vesicle formation and protein sorting: An Integrated Process. Annu. Rev. Biochem. 1997, 66, 511–548. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Woodman, E.; Lisanti, M. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Paraan, M.; Mendez, J.; Savanna, S.; Kurtin, D.; He, H.; Stagg, S.M. The structures of natively assembled clathrin-coated vesciles. Sci. Adv. 2020, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perrais, D.; Merrifield, C.J. Dynamics of Endocytic Vesicle Creation. Dev. Cell 2005, 9, 581–592. [Google Scholar] [CrossRef]
- Al-Thani, A.M.; Voss, S.C.; Al-Menhali, A.S.; Barcaru, A.; Horvatovich, P.; Jaganjac, M. Whole blood storage in CPDA1 blood bags alters erythrocyte membrane proteome. Oxidative Med. Cell. Longev. 2018, 10, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.M.; Chiu, D.T.; Yee, M.C.; Lubin, B.H. Red cell vsiculation—A common physiologic event. J. Lab. Clin. Med. 1986, 108, 315–324. [Google Scholar] [PubMed]
- Prudent, M.; Delobel, J.; Hübner, A.; Benay, C.; Lion, N.; Tissot, J.-D. Proteomics of Stored Red Blood Cell Membrane and Storage-Induced Microvesicles Reveals the Association of Flotillin-2 With Band 3 Complexes. Front. Physiol. 2018, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Bell, J.B.; Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Nitrite enhances hypoxic ATP synthesis and release of ATP into the vasculature: A new mechanism for nitrite-induced vesiculation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1494–H1503. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Liu, S.C.; Palek, J. Release of spectrin-free vesicles from human erythrocytes during ATP depletion: 1. characterization of spectrin-free vesicles. J. Cell Biol. 1977, 73, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Diebel, L.N.; Liberati, D.M. Red blood cell storage and adhesion to vascular endothelium under normal or stress conditions an in vitro microfluidic study. J. Trauma Acute Surg. 2019, 86, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Laurén, E.; Tigistu, F.; Valkonen, S.; Westberg, M.; Valkeäjarvi, A.; Eronen, J.; Siljanderde, P.; Pettilä, V.; Käkelä, R.; Laitinena, S.; et al. Phospholipid composition of packed red blood cells and that of extracellular vesicles show a high resemblence and stability during storage. Biochem. Biophys. Acta 2018, 1863, 1–8. [Google Scholar]
- Almizraq, R.J.; Holovati, J.L.; Acker, J.P. Characteristics of Extracellular Vesicles in Red Blood Concentrates Change with Storage Time and Blood Manufacturing Method. Transfus. Med. Hemother. 2018, 45, 185–193. [Google Scholar] [CrossRef]
- Levin, M.; Leppaanen, O.; Evaldsson, M.; Wiklund, O.; Bondjers, G.; Bjoornheden, T. Mapping of ATP, Glucose, Glycogen, and Lactate Concentrations Within the Arterial Wall. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1801–1807. [Google Scholar] [CrossRef][Green Version]
- Asaro, J.R.; Zhu, Q. Vital Erythrocyte phenomena: What can theory, modeling, and simulation offer? Biomech. Model. Mechanobiol. 2020, 19, 1361–1388. [Google Scholar] [CrossRef]
- Kuo, W.P.; Tigges, J.C.; Toxavidis, V.; Ghiran, I. Red Blood Cells: A Source of Extracellular Vesicles. Methods Mol. Biol. 2017, 1660, 15–22. [Google Scholar] [CrossRef]
- Safeukui, I.; Buffet, P.A.; Deplaine, G.; Perrot, S.; Brousse, V.; Ndour, A.; Nguyen, M.; Mercereau-Puijalon, O.; David, P.H.; Milon, G.; et al. Quantitative assessment of sensing and sequestration of spherocytic erythrocytes by the human spleen. Blood 2012, 120, 424–430. [Google Scholar] [CrossRef]
- Hochmuth, R.M.; Mohandas, N.; Blackshear, P.L. Measurement of the Elastic Modulus for Red Cell Membrane Using a Fluid Mechanical Technique. Biophys. J. 1973, 13, 747–762. [Google Scholar] [CrossRef]
- Berk, D.; Hochmuth, R. Lateral mobility of integral proteins in red blood cell tethers. Biophys. J. 1992, 61, 9–18. [Google Scholar] [CrossRef]
- Asaro, R.J.; Zhu, Q.; MacDonald, I.C. Tethering, evagination, and vesiculation via cell–cell interactions in microvascular flow. Biomech. Model. Mechanobiol. 2021, 20, 31–53. [Google Scholar] [CrossRef]
- Huisjes, R.; Bogdanova, A.; Van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; Van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwenhoek, A. Letter Dated March 26, 1675 to the Royal Society; Royal Society: London, UK, 1675. [Google Scholar]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mohandas, N.; Shobet, S.B. Osmotic gradient ektacytometry: Comprehensive characterization of red cell volume and surface naintenance. Blood 1983, 61, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Suner, L.; Galimand, J.; Bonnel, A.; Pascreau, T.; Couque, N.; Fenneteau, O.; Mohandas, N. Diagnostic tool for red blood cell membrane disorders: Assessment of a new generation ektacytometer. Blood Cells Mol. Dis. 2016, 56, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, E.; Gulbis, B.; Oirschot, B.; Wijk, R. Nex generation osmotic gradient ektacytometry for the diagnosis of hereditary spherocytosis: Interlaboratory method validation and experience. Clin. Chem. Lab. Med. 2017, 55, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Dobbe, J.G.G.; Streekstra, G.J.; Hardeman, M.R.; Ince, C.; Grimbergen, C.A. Measurement of the distribution of red blood cell deformability using an automated rheoscope. Cytometry 2002, 50, 313–325. [Google Scholar] [CrossRef]
- Shin, S.; Hou, J.X.; Suh, J.S.; Singh, M. Validation and application of a microfludic ektacytometer (RheoScan-D) in measuring erythrocyte deformability. Clin. Hemorrheol. Microcirc. 2007, 37, 319–328. [Google Scholar]
- Waugh, R.E.; Narla, M.; Jackson, C.W.; Mueller, T.J.; Suzuki, T.; Dale, G.I. Rheological properties of senescent erythrocytes: Loss of surface area and volume with red cell age. Blood 1992, 79, 1351–1358. [Google Scholar] [CrossRef]
- Sarkar, M.; Barari, S.K.; Mandai, D.B.; Nandankar, U.A.; Basu, A.; Mohanty, T.K.; Ray, S. The effect of anti-coagulants on the osmotic fragility of erythtocytes in the yak (Poephagus grunniens). Vet. J. 1999, 157, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Parpart, A.K.; Lorenz, P.B.; Parpart, E.R.; Gregg, J.R.; Chase, A.M. The osmotic resistance (fragility) of human red cells 1. J. Clin. Investig. 1947, 26, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Walski, T.; Chludzinska, L.; Komorowska, M.; Witkiewicz, W. Individual osmotic fragility distribution: A new parameter for determination of the osmotic properties of human red blood cells. BioMed Res. Int. 2014. [Google Scholar] [CrossRef]
- Knowles, D.W.; Tilley, L.; Mohandas, N.; Chasis, J.A. Erythrocyte membrane vesiculation: Model for the molecular mechanism of protein sorting. Proc. Natl. Acad. Sci. USA 1997, 94, 12969–12974. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Asaro, R.J.; Zhu, Q. Multiscale simulation of erythrocyte membranes. Phys. Rev. E 2010, 81, 031904. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.C.; Ragan, D.M.; Schmidt, E.E.; Groom, A.C. Kinetics of red blood cell passage through interendothelial slits into vevous sinuses in rat spleen, analyzed by in vivo microscopy. Microvasc. Res. 1987, 33, 118–134. [Google Scholar] [CrossRef]
- Tomishige, M.; Sako, Y.; Kusumi, A. Regulation Mechanism of the Lateral Diffusion of Band 3 in Erythrocyte Membranes by the Membrane Skeleton. J. Cell Biol. 1998, 142, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Kodippili, G.C.; Spector, J.; Sullivan, C.; Kuypers, F.A.; Labotka, R.; Gallagher, P.G.; Ritchie, K.; Low, P.S. Imaging of the diffusion of single band 3 molecules on normal and mutant erythrocytes. Blood 2009, 113, 6237–6245. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, H.G.; Rüpprl, D.A.; Galla, H.J.; Sackman, E. Lateral diffusion of lipids and glycophorin in solid phosphatidycholine bilayers. Biophys. J. 1984, 45, 577–587. [Google Scholar] [CrossRef]
- Lux, S.E. Anatomy of the red blood cell membrane skeleton: Unanswered questions. Blood 2015, 127, 187–199. [Google Scholar] [CrossRef]
- Drenckhahn, D.; Wagner, J. Stress fibers in the splenic sinud endothelium in situ: Molecular structure, relationship to extracellular matrix, and contractility. J. Cell Biol. 1986, 102, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.P.; Schnitter, M.G.H.; Seiffge, D.; Mittermayer, C. Induction of human vascular endothelial stress fibers by fluid shear stress. Nature 1984, 307, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef]
- Wang, J.H.C.; Goldschmidt-Clermont, P.; Yin, F.C.P. Contractility affects stress fiber remodeling abd reorientation of endothelial cells subjected to cyclic mechanical stretching. Ann. Biol. Eng. 2000, 28, 1165–1171. [Google Scholar] [CrossRef]
- Zhu, Q.; Salehyar, S.; Cabrales, P.; Asaro, R.J. Prospects for Human Erythrocyte Skeleton-Bilayer Dissociation during Splenic Flow. Biophys. J. 2017, 113, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Groom, A.C.; MacDonald, I.C.; Schmidt, E.E. Splenic microcirculatory blood flow and function with respect to red blood cells. In The Complete Speen; Bowdler, A.J., Ed.; Humanna Press: Totowa, NJ, USA, 2002; pp. 23–50. [Google Scholar]
- Schmidt, E.E.; Macdonald, I.C.; Groom, A.C. Microcirculatory pathways in normal human spleen, demonstrated by scanning electron microscopy of corrosion casts. Am. J. Anat. 1988, 181, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.; Schmidt, E.; Groom, A. The high splenic hematocrit: A rheological consequence of red cell flow through the reticular meshwork. Microvasc. Res. 1991, 42, 60–76. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Dailey, M.O. The immune function of the spleen. In The Complete Speen; Bowdler, A.J., Ed.; Humanna Press: Totowa, NJ, USA, 2002; pp. 51–69. [Google Scholar]
- Bishop, M.B.; Lansing, L.S. The spleen: A correlative overview of normal and pathologic anatomy. Hum. Pathol. 1982, 13, 334–342. [Google Scholar] [CrossRef]
- Chen, L.T.; Weiss, L. The role of the sinus wall in the passage of erythrocytes through the spleen. Blood 1973, 41, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, M.; Sirolli, V.; Gizza, F.; Stante, S.D.; Grilli, A.; Felaco, M. Enhanced adhesion of human uremic eruthrocytes to vascular endothelium: Role of phosphatidylserine exposure. Kidney Int. 2002, 62, 1358–1363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.-L.; Wautier, M.-P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin. Hemorheol. Microcirc. 2013, 53, 11–21. [Google Scholar] [CrossRef]
- Willekens, F.L.A.; Bregt-Roerdinkholder, B.; Groenen-döpp, Y.A.; Bos, H.J.; Bosman, G.J.C.G.M.; Bos, A.G.v.; Verkeij, A.J.; Weere, J.M. Haemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood 2003, 101, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Oberieithner, H.; White, M.; Kusche-Vihrog, K. Sodium renders endothelial cells sticky for red blood cells. Front. Physiol. 2015, 6, 188. [Google Scholar]
- Fujita, T. A Scanning Electron Microscope Study of the Human Spleen. Arch. Histol. Jpn. 1974, 37, 187–216. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, R.M.; Marcus, D. Membrane tethers formed from blood cells with available area and determination of the adhesion energy. Biophys. J. 2002, 82, 2964–2969. [Google Scholar] [CrossRef]
- Hochmuth, R.; Evans, E. Extensional flow of erythrocyte membrane from cell body to elastic tether. I. Analysis. Biophys. J. 1982, 39, 71–81. [Google Scholar] [CrossRef]
- Hochmuth, R.; Wiles, H.; Evans, E.; McCown, J. Extensional flow of erythrocyte membrane from cell body to elastic tether. II. Experiment. Biophys. J. 1982, 39, 83–89. [Google Scholar] [CrossRef]
- Dai, J.; Sheetz, M.P. Membrane tether formation fron blebbing cells. Biophys. J. 1999, 77, 3363–3370. [Google Scholar] [CrossRef]
- Hosu, B.G.; Sun, M.; Marga, F.; Grandbois, M.; Forgacs, G. Eukaryotic membrane tethers revisited using magnetic tweezers. Phys. Biol. 2007, 4, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Borghi, N.; Brochard-Wyart, F. Tether Extrusion from Red Blood Cells: Integral Proteins Unbinding from Cytoskeleton. Biophys. J. 2007, 93, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.; Waugh, R. Energy of dissociation of lipid bilayer from the membrane skeleton of red blood cells. Biophys. J. 1997, 72, 2669–2678. [Google Scholar] [CrossRef]
- Pernow, J.; Mahdi, A.; Yang, J.; Zhou, Z. Red blood cell dysfunction: A new player in cardiovascular disease. Cardiovasc. Res. 2019, 115, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Colin, Y.; Le Van Kim, C.; El Nemer, W. Red cell adhesion in human diseases. Curr. Opin. Hematol. 2014, 21, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Kucukal, E.; Ilich, A.; Key, N.S.; Little, J.A.; Gurkan, U.A. Red blood cell adhesion to heme-activated endothelial cells reflects clinical phenotype in sickle cell disease. Am. J. Hematol. 2018, 93, 1050–1060. [Google Scholar] [CrossRef]
- Kucukal, E.; Man, Y.; Quinn, E.; Tewari, N.; An, R.; Ilich, A.; Key, N.S.; Little, J.A.; Gurkan, U.A. Red blood cell adhesion to ICAM-1 is mediated by fibrinogen and is associated with right-to-left shunts in sickle cell disease. Blood Adv. 2020, 4, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D. Sickle red cell adhesion: Many issues and some answers. Transfus. Clin. Biol. 2008, 15, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Jambou, R.; Combes, V.; Jambou, M.-J.; Weksler, B.B.; Couraud, P.-O.; Grau, G.E. Plasmodium falciparum adhesion on human brain microvascular endolethial cells involves transmigration-like cup formation and induces opeing of intercellular junctions. PLoS Pathog. 2010, 6, e1001021. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.A.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Asaro, R.J.; Lin, K.; Zhu, Q. Mechanosensitivity Occurs along the Adhesome’s Force Train and Affects Traction Stress. Biophys. J. 2019, 117, 1599–1614. [Google Scholar] [CrossRef] [PubMed]
- Kucukal, E.; Little, J.A.; Gurkan, U.A. Shear dependent red blood cell adhesion in microscale flow. Integr. Biol. 2018, 10, 194–206. [Google Scholar] [CrossRef]
- Bosman, G.F.; Willekens, L.A.; Weere, J.M. Erythocyte Senesecence. In Erythrocytes, Physiology and Pathophysiology; Land, F.M., Föller, M., Eds.; Imperial College Press: London, UK, 2012. [Google Scholar]
- Davidson, M.B.; Schriger, D.L. Effect of age and race/ethnicity on HbA1c levels in people without known diabetes mellitus: Implications for the diagnosis of diabetes. Diabetes Res. Clin. Pract. 2010, 87, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Paton, R.C.; Wautier, M.P.; Pintigny, D.; Abadie, E.; Passa, P.; Caen, J.P. Increased adhesion of erythrocytes to endothelial cells in diabetes mellitus and its relation to vascular complications. N. Engl. J. Med. 1981, 305, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Hori, O.; Cao, R.; Yan, S.D.; Brett, J.; Wautier, J.L.; Ogawa, S.; Kuwabara, K.; Matsumoto, M.; Stern, D. RAGE: A novel cellular receptor for advanced glycation end products. Diabetes 1996, 45, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor—Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fang, W.; Ma, L.; Wang, Z.-D.; Yang, Y.-M.; Lu, Y.-Q. VEGF levels in plasma in relation to metabolic control, inflammation, and microvascular complications in type-2 diabetes. Medicine 2018, 97, e0415. [Google Scholar] [CrossRef]
- Setty, Y.B.; Betal, S.G. Microvascular Endothelial Cells Express Phosphatidylserine Receptor: A Functionally Active Receptor for Phosphatidylserine-Positive Erythrocytes. Blood 2007, 110, 1720. [Google Scholar] [CrossRef]
- Campa, C.; Alivernini, G.; Bolletta, E.; Parodi, M.; Perri, P. Anti-VEGF Therapy for Retinal vien occlusion. Curr. Drug Targets 2016, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [CrossRef]
- Gutsaeva, D.R.; Montero-Huerta, P.; Parkerson, J.B.; Yerigenahally, S.D.; Ikuta, T.; Head, C.A. Molecular mechanisms underlying synergistic adhesion of sickle red blood cells by hypoxia and low nitric oxcide bioavailability. Blood 2014, 123, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Trinh-Trang-Tan, M.-M.; Vilela-Lamego, C.; Picot, J.; Wautier, M.-P.; Cartron, J.-P. Intercellular adhesion molecule-4 and CD36 are implicated in the abnormal adhesiveness of sickle cell SAD mouse erythrocytes to endothelium. Haematologica 2009, 95, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Fel, A.; Lewandowska, A.E.; Petrides, P.E.; Wiśniewski, J.R. Comparison of protein composition of serum enriched in extracellular vesicles isolated from polycythemia vera patients and healthy controls. Proteomes 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.-M.; Toti, F. Cellular Mechanisms Underlying the Formation of Circulating Microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferive neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, J. Electrokinetic effect of the endothelial endothelial glycocalyx layer on two-phase blood flow in small vlood vessels. Microvasc. Res. 2009, 78, 14–19. [Google Scholar] [CrossRef]
- Lipowsky, H.; Gao, L.; Lescanic, A. Shedding of the endothelial glycocalyx in aterioles capillaries and venules and its effect on capillary hemodynamics during inflammation. Am. J. Physiol. 2011, 301, 2235–2245. [Google Scholar]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef]
- Noble, M.I.; Drake-Holland, A.J.; Vink, H. Hypothesis: Arterial glycocalyx dysfuction is the first step in the atherothrombotic process. QJM 2008, 101, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Neu, B.; Sowemimo-Coker, S.O.; Meiselman, H.J. Cell-Cell Affinity of Senescent Human Erythrocytes. Biophys. J. 2003, 85, 75–84. [Google Scholar] [CrossRef]
- Beutler, E.; Lichman, M.A.; Coller, B.S.; Kipps, T.J. Hemotology; McGraw Hill: New York, NY, USA, 1995. [Google Scholar]
- Arosa, F.A.; Pereira, C.F.; Fonseca, A.M. Red blood cells as modulators of T cell growth and survivial. Curr. Pharm. Des. 2004, 10, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Danesh, A.; Inglis, H.C.; Jackman, R.P.; Wu, S.; Deng, X.; Muench, M.O.; Heitman, J.W.; Norris, P.J. Exosomes from red blood cell units bind to monocytes and induce preinflammatory cytokines, boosting T-cell responses in vitro. Blood 2014, 123, 687–696. [Google Scholar] [CrossRef]
- Steinman, R.M. The dentritic cell system and its role in immunogenicity. Ann. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J.C.G.M.; Lasonder, E.; Luten, M.; Roerdinkholder-Stoelwinder, B.; Novotný, V.M.J.; Bos, H.; Grip, W.J.D. The proteome of red cell membranes and vesicles during storage in blood bank conditions. Transfusion 2008, 48, 827–835. [Google Scholar]
- Schäkel, K.; von Kietzell, M.; Hänsel, A.; Ebling, A.; Schulze, L.; Haase, M.; Semmler, C.; Sarfati, M.; Barclay, A.N.; Randolph, G.J.; et al. Human 6-sulfo LacNAc-expressing dentritic cells are principal producers of early interleukin-12 and are controlled by erythrocytes. Immunity 2006, 24, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Hogman, C.F.; Meryman, H.T. Storage parameters affecting red cell survivial and function after transfusion. Transfus. Med. Rev. 1999, 13, 275–296. [Google Scholar] [CrossRef]
- Leppänen, O.; Björnheden, T.; Evaldsson, M.; Borén, J.; Wiklund, O.; Levin, M. ATP depletion in macrophages in the core of advanced rabbit atherosclerotic plaques in vivo. Atherosclerosis 2006, 188, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Rifkind, J.M. Reaction of hydrogen peroxide with ferrymemoglobin: Superoxide production and heme degradation. Biochemistry 2000, 39, 12503–12511. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S.; Waugh, S.M.; Zinke, K.; Drenckhahn, D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science 1985, 227, 531–533. [Google Scholar] [CrossRef]
- Nomura, S.; Suzuki, M.; Katsura, K.; Xie, G.L.; Miyazaki, Y.; Miyake, T.; Kido, H.; Kagawa, H.; Fukuhara, S. Platelet-derived vesicles may influence the development of atherosclerosis in diabetes mellitus. Atherosclerosis 1995, 116, 235–240. [Google Scholar] [CrossRef]
- Tziakas, D.N.; Chalikas, G.K.; Stakos, D.; Boudolas, H. The role of red blood cells in the progression and instability of atherosclerotic plaque. Int. J. Cardiol. 2010, 142, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.; Chalikias, G.; Kapelouzou, A.; Tentes, I.; Schäfer, K.; Karayannakos, P.; Kostakis, A.; Boudoulas, H.; Konstantinides, S. Erythrocyte membrane cholesterol and lipid core growth in a rabbit model of atherosclerosis: Modulatory effects of rosuvastatin. Int. J. Cardiol. 2013, 170, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F.; Arias, C.F. How do red blood cells know when to die? R. Soc. Open Sci. 2017, 4, 160850. [Google Scholar] [CrossRef]
- da Silveira Cavalcante, L.; Acker, J.P.; Holovati, J.L. Differences in Rat and Human Erythrocytes Following Blood Component Manufacturing: The Effect of Additive Solutions. Transfus. Med. Hemother. 2015, 42, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Lutz, U.H.; Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 2013, 4, 1–15. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Topez, O.; Cohen, L.A.; Yakov, L.D.; Haber, T.; Morgenstern, A.; Weiss, A.; Berman, K.C.; Fibach, E.; Meyron-Holtz, E. Physiological aged red blood cells undergo erythrophagocytosis in vivo but not in vitro. Haematologica 2012, 97, 994–1002. [Google Scholar] [CrossRef]
- Durocher, J.R.; Payne, R.C.; Conrad, M.E. Role of sialic acid in erythrocyte survival. Blood 1975, 45, 11–20. [Google Scholar] [CrossRef]
- Jancik, J.; Schauer, R. Sialic Acid—A Determinant of the Life-Time of Rabbit Erythrocytes. Biol. Chem. 1974, 355, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Jancik, J.; Schauer, R.; Streicher, H.J. Influence of membrane-bound N-acetylneuraminic acid on the survival of erythrocytes in man. Hoppe Seyler’s Z. Physiol. Chem. 1975, 356, 1329–1331. [Google Scholar]
- Bocci, V. The role of sialic acid in determining the life-span of circulating cells and glycoproteins. Cell. Mol. Life Sci. 1976, 32, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Nigam, P.K.; Narain, V.S.; Kumar, A. Sialic acid in cardiovascular diseases. Indian J. Clin. Biochem. 2006, 21, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Saha, S.; Basu, S.; Chakrabarti, A. Porous red cell ultrastructure and loss of membrane asymmetry in a novel case of hemolytic anemia. Eur. J. Haematol. 2008, 81, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Bratosin, D.; Mazurier, J.; Debray, H.; Lecocq, M.; Boilly, B.; Alonso, C.; Moisei, M.; Motas, C.; Montreuil, J. Flow cytofluorimetric analysis of young and senescent human erythrocytes probed with lectins. Evidence that sialic acids control their life span. Glycoconj. J. 1995, 12, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Hadengue, A.L.; Del-Pino, M.; Simon, A.; Levenson, J. Erythrocyte Disaggregation Shear Stress, Sialic Acid, and Cell Aging in Humans. Hypertension 1998, 32, 324–330. [Google Scholar] [CrossRef]
- Yang, Y.; Koo, S.; Lin, C.S.; Neu, B. Specific bonding of red blood cells to endothelial cells is regulated by nonadsorbing macromolecules. J. Biol. Chem. 2010, 285, 40489–40495. [Google Scholar] [CrossRef] [PubMed]
- Alapan, Y.; Little, J.A.; Gurkan, U.A. Heterogeneous Red Blood Cell Adhesion and Deformability in Sickle Cell Disease. Sci. Rep. 2015, 4, 7173. [Google Scholar] [CrossRef]
- Tsvirkun, D.; Grichine, A.; Duperray, A.; Misbah, C.; Bureau, L. Microvasculature on a chip: Study of the Endothelial Surface Layer and the flow structure of Red Blood Cells. Sci. Rep. 2017, 7, srep45036. [Google Scholar] [CrossRef]
- Sztilovics, M.; Gerecsel, T.; Peter, B.; Saftics, A.; Kurunczi, S.; Szekacs, I.; Szabó, B.; Horvath, R. Single-cell adhesion force kinetics of cell populations from combined label-free optical biosensors and robotic fluid force microscopy. Sci. Rep. 2020, 10, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Orgovan, N.; Peter, B.; Bäsze, S.; Ramsden, J.J.; Szabó, B.; Horvath, R. Dependence of cancer cell adhesion kinetics on integrin ligand surface density measured by a high-throughput label-free resonant waveguide grating biosensor. Sci. Rep. 2014, 4, 4034–4041. [Google Scholar] [CrossRef] [PubMed]
- Ermis, M.; Antmen, E.; Hasirci, V. Micro and Nanofabrication methods to control cell-substrate interactions and cell behavior: A review from the tissue engineering perspective. Bioact. Mater. 2018, 3, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Ciana, A.; Achilli, C.; Minetti, G. Membrane rafts of the human red blood cell. Mol. Membr. Biol. 2014, 31, 47–57. [Google Scholar] [CrossRef]
- Drzeniek, R. Differences in splitting capacity of virus and V. cholerae neuraminidases on sialic acid type substrates. Biochem. Biophys. Res. Commun. 1967, 26, 631–638. [Google Scholar] [CrossRef]
- Gilson, C.R.; Kraus, T.S.; Hod, E.A.; Hendrickson, J.E.; Spitalnik, S.L.; Hillyer, C.D.; Shaz, B.H.; Zimring, J.C. A novel mouse model of red bllod cell storage and posttransfusion in vivo survival. Transfusion 2009, 49, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Straat, M.; Klei, T.; De Korte, D.; Van Bruggen, R.; Juffermans, N.P. Accelerated clearance of human red blood cells in a rat transfusion model. Intensive Care Med. Exp. 2015, 3, 27. [Google Scholar] [CrossRef]
- Hod, E.A.; Arinsburg, S.A.; Francis, R.O.; Hendrickson, J.E.; Zimring, J.C.; Spitalnik, S.L. Use of mouse models to study the mechanisms and consequences of RBC clearance. Vox Sang. 2010, 99, 99–111. [Google Scholar] [CrossRef]
- Kyriakou, D.S.; Alexandrakis, M.G.; Kyriakou, E.S.; Kourelis, D.L.T.V.; Passam, P.; Papadakis, A. Activated peripheral blood and endothelial cells in thalassemia patients. Ann. Hematol. 2001, 80, 577–583. [Google Scholar] [PubMed]
- Sorensen, E.W.; Lian, J.; Ozga, A.J.; Miyabe, Y.; Ji, S.W.; Bromley, S.K.; Mempel, T.R.; Luster, A.D. CXCL10 stabilizes T cell-brain endothelial cell adhesion leading to the introduction of cerebral malaria. JCI Insight 2018, 3, e98911. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, A.; Engler, A. Acute shear stress direction dictates adherent cell remodeling and verifies shear profile of spinning disc assays. Phys. Biol. 2015, 12, 016011. [Google Scholar] [CrossRef] [PubMed]
- Kucukal, E.; Man, Y.; Hill, A.; Liu, S.; Bode, A.; An, R.; Kadambi, J.; Little, J.A.; Gurkan, U.A. Whole blood viscosity and red cell adhesion: Potential biomarkers for targeted and curative therapies in sickel cell disease. Am J. Hematol. 2020, 95, 1246–1256. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asaro, R.J.; Cabrales, P. Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow. Diagnostics 2021, 11, 971. https://doi.org/10.3390/diagnostics11060971
Asaro RJ, Cabrales P. Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow. Diagnostics. 2021; 11(6):971. https://doi.org/10.3390/diagnostics11060971
Chicago/Turabian StyleAsaro, Robert J., and Pedro Cabrales. 2021. "Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow" Diagnostics 11, no. 6: 971. https://doi.org/10.3390/diagnostics11060971
APA StyleAsaro, R. J., & Cabrales, P. (2021). Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow. Diagnostics, 11(6), 971. https://doi.org/10.3390/diagnostics11060971