Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions



Abstract
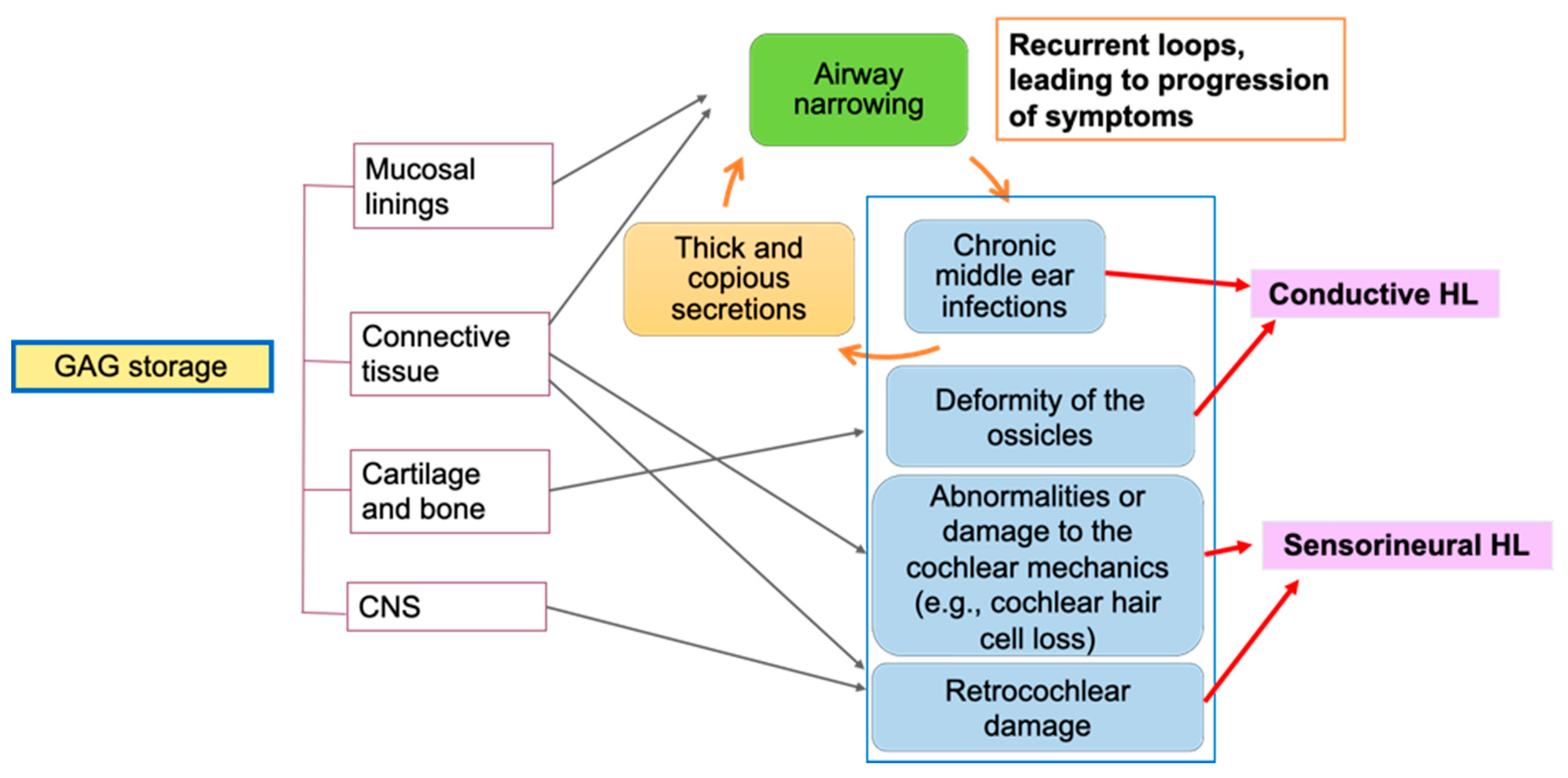
1. Introduction
2. Mucopolysaccharidosis Type I (Hurler Syndrome)
2.1. Types and Cause of Hearing Loss
2.2. Efficacy of Clinical Treatments on Hearing
3. Mucopolysaccharidosis Type II (Hunter Syndrome)
3.1. Audiological Findings
3.2. Causes of Hearing Loss
3.3. Efficacy of Clinical Treatments on Hearing
4. Mucopolysaccharidosis Type III (Sanfilippo Syndrome)
4.1. Audiological Findings
4.2. Cause of Hearing Loss
4.3. Efficacy of Clinical Treatments on Hearing
5. Mucopolysaccharidosis Type IV (Morquio Syndrome)
5.1. Hearing Loss
5.2. Efficacy of Clinical Treatments on Hearing
6. Mucopolysaccharidosis Type VI (Maroteaux–Lamy Syndrome)
6.1. Hearing Loss in MPS VI
6.2. Efficacy of Clinical Treatment on Hearing
7. Mucopolysaccharidosis Type VII (Sly Syndrome)
7.1. Causes of Hearing Loss
7.2. Audiological Findings
7.3. Efficacy of Clinical Treatment on Hearing
8. Mucopolysaccharidosis Type IX (Natowicz Syndrome)
8.1. Hearing Issues
8.2. Animal Models
9. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MPS | Mucopolysaccharidosi(e)s |
| GAGs | Glycosaminoglycans |
| ERT | Enzyme replacement therapy |
| dB HL | dB hearing level |
| DS | Dermatan sulfate |
| HS | Heparan sulfate |
| HSCT | Hematopoietic stem cell transplantation |
| OAE | Otoacoustic emissions |
| ABR | Auditory brainstem response |
| CNS | Central nervous system |
| BMT | Bone marrow transplantation |
| AAV | Adeno-associated virus |
| KS | Keratan sulfate |
| DPOAE | Distortion products otoacoustic emissions |
| ARSB | Arylsulfatase B |
| HYAL1 | Hyaluronidase 1 |
| HYAL3 | Hyaluronidase 3 |
| HYAL2 | Hyaluronidase 2 |
| HL | Hearing loss |
References
- Wraith, J.E.; Rogers, J.G.; Danks, D.M. The mucopolysaccharidoses. J. Paediatr. Child. Health 1987, 23, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Demydchuk, M.; Hill, C.H.; Zhou, A.; Bunkóczi, G.; Stein, P.E.; Marchesan, D.; Deane, J.E.; Read, R.J. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat. Commun. 2017, 8, 15786. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.; Aldámiz-Echevarría, L.; Llarena, M.; Couce, M.L. Sanfilippo syndrome: Overall review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Peracha, H.; Sawamoto, K.; Averill, L.; Kecskemethy, H.; Theroux, M.; Thacker, M.; Nagao, K.; Pizarro, C.; Mackenzie, W.; Kobayashi, H.; et al. Molecular genetics and metabolism, special edition: Diagnosis, Diagnosis and prognosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Tomatsu, S.; Mason, R.W.; Yasuda, E.; Mackenzie, W.G.; Hossain, J.; Shibata, Y.; Montaño, A.M.; Kubaski, F.; Giugliani, R.; et al. Di-sulfated Keratan Sulfate as a Novel Biomarker for Mucopolysaccharidosis II, IVA, and IVB. JIMD Rep. 2015, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tomanin, R.; Karageorgos, L.; Zanetti, A.; Al-Sayed, M.; Bailey, M.; Miller, N.; Sakuraba, H.; Hopwood, J.J. Mucopolysaccharidosis type VI (MPS VI) and molecular analysis: Review and classification of published variants in the ARSB gene. Hum. Mutat. 2018, 39, 1788–1802. [Google Scholar] [CrossRef]
- Natowicz, M.R.; Short, M.P.; Wang, Y.; Dickersin, G.R.; Gebhardt, M.C.; Rosenthal, D.I.; Sims, K.B.; Rosenberg, A.E. Clinical and biochemical manifestations of hyaluronidase deficiency. N. Engl. J. Med. 1996, 335, 1029–1033. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef]
- Simmons, M.A.; Bruce, I.A.; Penney, S.; Wraith, E.; Rothera, M.P. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 589–595. [Google Scholar] [CrossRef]
- Silveira, M.R.; Buriti, A.K.L.; Martins, A.M.; Gil, D.; Azevedo, M.F. Audiometric evaluation in individuals with mucopolysaccharidosis. Clin. Sao Paulo 2018, 73, e523. [Google Scholar] [CrossRef]
- Lenka, M.; Michal, J.; Pavel, J.; Vera, M.; Marketa, B.; Jiri, Z.; Martin, M. Otorhinolaryngological manifestations in 61 patients with mucopolysaccharidosis. Int. J. Pediatr. Otorhinolaryngol. 2020, 135, 110137. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, J.J.; Park, S.-I.; Cho, S.-Y.; Jin, D.-K.; Cho, Y.-S.; Chung, W.-H.; Hong, S.-H.; Moon, I.J. Auditory Characteristics in Patients With Mucopolysaccharidosis. Otol. Neurotol. Off. Publ. Am. Otol. Soc. Am. Neurotol. Soc. Eur. Acad. Otol. Neurotol. 2019, 40, e955–e961. [Google Scholar] [CrossRef] [PubMed]
- Mesolella, M.; Cimmino, M.; Cantone, E.; Marino, A.; Cozzolino, M.; Della Casa, R.; Parenti, G.; Iengo, M. Management of otolaryngological manifestations in mucopolysaccharidoses: Our experience. Acta Otorhinolaryngol. Ital. Organo Uff. Della Soc. Ital. Otorinolaringol. E Chir. Cerv.-Facc. 2013, 33, 267–272. [Google Scholar]
- Lin, H.Y.; Shih, S.C.; Chuang, C.K.; Lee, K.S.; Chen, M.R.; Lin, H.C.; Chiu, P.C.; Niu, D.M.; Lin, S.P. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 533–538. [Google Scholar] [CrossRef]
- Vargas-Gamarra, M.F.; de Paula-Vernetta, C.; Vitoria Minana, I.; Ibanez-Alcaniz, I.; Cavalle-Garrido, L.; Alamar-Velazquez, A. Audiological findings in children with mucopolysaccharidoses type i-iv. Acta Otorrinolaringol. Esp. 2017, 68, 262–268. [Google Scholar] [CrossRef]
- Gokdogan, C.; Altinyay, S.; Gokdogan, O.; Tutar, H.; Gunduz, B.; Okur, I.; Tumer, L.; Kemaloglu, Y.K. Audiologic evaluations of children with mucopolysaccharidosis. Braz. J. Otorhinolaryngol. 2016, 82, 281–284. [Google Scholar] [CrossRef]
- Giraldo, L.J.M.; Arturo-Terranova, D.; Soto, J.M.S. Otorhinolaryngological Findings in Patients from Southwestern Colombia with Clinical, Enzymatic and Molecular Diagnosis of Mucopolysaccharidosis II, IV-A and VI. J. Inborn Errors Metab. Screen. 2020, 8, e20190006. [Google Scholar] [CrossRef]
- Da Costa, V.; O’Grady, G.; Jackson, L.; Kaylie, D.; Raynor, E. Improvements in Sensorineural Hearing Loss After Cord Blood Transplant in Patients With Mucopolysaccharidosis. Arch. Otolaryngol. Neck Surg. 2012, 138, 1071–1076. [Google Scholar] [CrossRef][Green Version]
- van den Broek, B.T.A.; Smit, A.L.; Boelens, J.J.; van Hasselt, P.M. Hearing loss in patients with mucopolysaccharidoses-1 and -6 after hematopoietic cell transplantation: A longitudinal analysis. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Dualibi, A.P.; Martins, A.M.; Moreira, G.A.; de Azevedo, M.F.; Fujita, R.R.; Pignatari, S.S.N. The impact of laronidase treatment in otolaryngological manifestations of patients with mucopolysaccharidosis. Braz. J. Otorhinolaryngol. 2016, 82, 522–528. [Google Scholar] [CrossRef]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, I.; Spellacy, E.; Crow, J.; Watts, R.W.E. Histopathological studies of the temporal bones in Hurler’s disease [mucopolysaccharidosis (MPS) IH]. J. Laryngol. Otol. 1985, 99, 29–41. [Google Scholar] [CrossRef]
- Kariya, S.; Schachern, P.A.; Nishizaki, K.; Paparella, M.M.; Cureoglu, S. Inner ear changes in mucopolysaccharidosis type I/Hurler syndrome. Otol. Neurotol. 2012, 33, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Souillet, G.; Guffon, N.; Maire, I.; Pujol, M.; Taylor, P.; Sevin, F.; Bleyzac, N.; Mulier, C.; Durin, A.; Kebaili, K.; et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003, 31, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Schachern, P.A.; Cureoglu, S.; Tsuprun, V.; Paparella, M.M.; Whitley, C.B. Age-related functional and histopathological changes of the ear in the MPS I mouse. Int. J. Pediatr. Otorhinolaryngol. 2007, 71, 197–203. [Google Scholar] [CrossRef][Green Version]
- Schachern, P.A.; Shea, D.A.; Paparella, M.M. Mucopolysaccharidosis I-H (Hurler’s syndrome) and human temporal bone histopathology. Ann. Otol. Rhinol. Laryngol. 1984, 93, 65–69. [Google Scholar] [CrossRef]
- Keilmann, A.; Nakarat, T.; Bruce, I.A.; Molter, D.; Malm, G.; HOS Investigators. Hearing loss in patients with mucopolysaccharidosis II: Data from HOS-the Hunter Outcome Survey. J. Inherit. Metab. Dis. 2012, 35, 343–353. [Google Scholar] [CrossRef]
- Chiong, M.A.D.; Canson, D.M.; Abacan, M.A.R.; Baluyot, M.M.P.; Cordero, C.P.; Silao, C.L.T. Clinical, biochemical and molecular characteristics of Filipino patients with mucopolysaccharidosis type II-Hunter syndrome. Orphanet J. Rare Dis. 2017, 12, 7. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Hong, S.H.; Chu, H.; Kim, K.R.; Ko, M.H.; Kwon, S.Y.; Moon, I.J.; Chung, W.H.; Cho, Y.S.; Kim, C.H.; Suh, M.W.; et al. Auditory characteristics and therapeutic effects of enzyme replacement in mouse model of the mucopolysaccharidosis (MPS) II. Am. J. Med. Genet. Part A 2012, 158, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, D.; Thakkar, K.; Poe, M.; Escolar, M.L. Natural history of Sanfilippo syndrome type A. J. Inherit. Metab. Dis. 2014, 37, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Heldermon, C.D.; Hennig, A.K.; Ohlemiller, K.K.; Ogilvie, J.M.; Herzog, E.D.; Breidenbach, A.; Vogler, C.; Wozniak, D.F.; Sands, M.S. Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS ONE 2007, 2, e772. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Morlet, T.; Haley, E.; Padilla, J.; Nemith, J.; Mason, R.W.; Tomatsu, S. Neurophysiology of hearing in patients with mucopolysaccharidosis type IV. Mol. Genet. Metab. 2018, 123, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Riedner, E.D.; Levin, L.S. Hearing patterns in Morquio’s syndrome (mucopolysaccharidosis IV). Arch. Otolaryngol. 1977, 103, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Swartz, D.J.; Santi, P.A. Immunohistochemical localization of keratan sulfate in the chinchilla inner ear. Hear. Res. 1997, 109, 92–101. [Google Scholar] [CrossRef]
- Gomes, D.F.; Gallo, L.G.; Leite, B.F.; Silva, R.B.; da Silva, E.N. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: Systematic review. J. Inherit. Metab. Dis. 2019, 42, 66–76. [Google Scholar] [CrossRef]
- Furujo, M.; Kosuga, M.; Okuyama, T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI: 10-Year follow up. Mol. Genet. Metab. Rep. 2017, 13, 69–75. [Google Scholar] [CrossRef]
- Harmatz, P.; Garcia, P.; Guffon, N.; Randolph, L.M.; Shediac, R.; Braunlin, E.; Lachman, R.S.; Decker, C. Galsulfase (Naglazyme(R)) therapy in infants with mucopolysaccharidosis VI. J. Inherit. Metab. Dis. 2014, 37, 277–287. [Google Scholar] [CrossRef]
- Horovitz, D.D.G.; Magalhães, T.S.P.C.; Acosta, A.; Ribeiro, E.M.; Giuliani, L.R.; Palhares, D.B.; Kim, C.A.; de Paula, A.C.; Kerstenestzy, M.; Pianovski, M.A.D.; et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol. Genet. Metab. 2013, 109, 62–69. [Google Scholar] [CrossRef]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Çoker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Ohlemiller, K.K.; Hennig, A.K.; Lett, J.M.; Heidbreder, A.F.; Sands, M.S. Inner ear pathology in the mucopolysaccharidosis VII mouse. Hear. Res. 2002, 169, 69–84. [Google Scholar] [CrossRef]
- O’Connor, L.H.; Erway, L.C.; Vogler, C.A.; Sly, W.S.; Nicholes, A.; Grubb, J.; Holmberg, S.W.; Levy, B.; Sands, M.S. Enzyme replacement therapy for murine mucopolysaccharidosis type VII leads to improvements in behavior and auditory function. J. Clin. Investig. 1998, 101, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.S.; Erway, L.C.; Vogler, C.; Sly, W.S.; Birkenmeier, E.H. Syngeneic bone marrow transplantation reduces the hearing loss associated with murine mucopolysaccharidosis type VII. Blood 1995, 86, 2033–2040. [Google Scholar] [CrossRef]
- Berry, C.L.; Vogler, C.; Galvin, N.J.; Birkenmeier, E.H.; Sly, W.S. Pathology of the ear in murine mucopolysaccharidosis type VII. Morphologic correlates of hearing loss. Lab. Investig. J. Tech. Methods Pathol. 1994, 71, 438–445. [Google Scholar]
- Imundo, L.; Leduc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Matte, U.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagn. Basel Switz. 2020, 10, 161. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Giugliani, R.; Harmatz, P.; Lin, S.-P.; Scarpa, M. Assessing the impact of the five senses on quality of life in mucopolysaccharidoses. Orphanet J. Rare Dis. 2020, 15, 97. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Jarnes, J.; Ahmed, A.; Nestrasil, I.; Ziegler, R.; Delaney, K.; Shapiro, E.; Whitley, C. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol. Genet. Metab. Rep. 2017, 13, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Laraway, S.; Breen, C.; Mercer, J.; Jones, S.; Wraith, J.E. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Mol. Genet. Metab. 2013, 109, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef] [PubMed]
- Tokic, V.; Barisic, I.; Huzjak, N.; Petkovic, G.; Fumic, K.; Paschke, E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur. J. Pediatr. 2006, 166, 727. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Navarro, E.; Domingo-Jiménez, M.R.; Alcalde-Martín, C.; Cancho-Candela, R.; Couce, M.L.; Galán-Gómez, E.; Alonso-Luengo, O. Clinical manifestations in female carriers of mucopolysaccharidosis type II: A Spanish cross-sectional study. Orphanet J. Rare Dis. 2013, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and Diagnosis of Mucopolysaccharidosis II (Hunter Syndrome). Pediatrics 2008, 121, e377. [Google Scholar] [CrossRef]
- Bicalho, C.G.; Rezende, M.M.; Nogueira, A.M.C.M.; Paulon, R.M.C.; Acosta, A.X. The importance of the otorhinolaryngologic evaluation in mucopolysaccharidosis patients. Int. Arch. Otorhinolaryngol. 2011, 15, 290–294. [Google Scholar] [CrossRef]
- Walker, R.; Belani, K.G.; Braunlin, E.A.; Bruce, I.A.; Hack, H.; Harmatz, P.R.; Jones, S.; Rowe, R.; Solanki, G.A.; Valdemarsson, B. Anaesthesia and airway management in mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 211–219. [Google Scholar] [CrossRef]
- Borgo, A.; Cossio, A.; Gallone, D.; Vittoria, F.; Carbone, M. Orthopaedic challenges for mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 123. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatric Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, J.Y.; Choo, O.-S.; Jang, J.H.; Park, H.Y.; Choung, Y.-H. Cochlear Implantation via the Transmeatal Approach in an Adolescent with Hunter Syndrome-Type II Mucopolysaccharidosis. J. Audiol. Otol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bartsocas, C.; Gröbe, H.; van de Kamp, J.J.; von Figura, K.; Kresse, H.; Klein, U.; Giesberts, M.A. Sanfilippo type C disease: Clinical findings in four patients with a new variant of mucopolysaccharidosis III. Eur. J. Pediatr. 1979, 130, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.M.; Cao, H.; Kaplan, P.; Silver, K.; Leonard, G.; De Meirleir, L.; Lissens, W.; Liebaers, I.; Veilleux, M.; Andermann, F.; et al. Sanfilippo Syndrome Type D: Natural History and Identification of 3 Novel Mutations in the GNS Gene. Arch. Neurol. 2007, 64, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Chuang, C.-K.; Lee, C.-L.; Tu, R.-Y.; Lo, Y.-T.; Chiu, P.C.; Niu, D.-M.; Fang, Y.-Y.; Chen, T.-L.; Tsai, F.-J.; et al. Mucopolysaccharidosis III in Taiwan: Natural history, clinical and molecular characteristics of 28 patients diagnosed during a 21-year period. Am. J. Med. Genet. Part A 2018, 176, 1799–1809. [Google Scholar] [CrossRef]
- Zelei, T.; Csetneki, K.; Vokó, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef]
- Ruijter, G.J.G.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef]
- Valstar, M.J.; Bertoli-Avella, A.M.; Wessels, M.W.; Ruijter, G.J.G.; de Graaf, B.; Olmer, R.; Elfferich, P.; Neijs, S.; Kariminejad, R.; Suheyl Ezgü, F.; et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutationsb. Hum. Mutat. 2010, 31, E1348–E1360. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Savvopoulou-Augoustidou, P.A.; Sewell, A.; Papadopoulou, F.; Badouraki, M.; Vargiami, E.; Gombakis, N.P.; Katzos, G.S. Serial magnetic resonance imaging findings in mucopolysaccharidosis IIIB (Sanfilippo’s syndrome B). Brain Dev. 2001, 23, 385–389. [Google Scholar] [CrossRef]
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Węgrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1–10. [Google Scholar] [CrossRef]
- Lau, A.A.; Shamsani, N.J.; Winner, L.K.; Hassiotis, S.; King, B.M.; Hopwood, J.J.; Hemsley, K.M. Neonatal Bone Marrow Transplantation in MPS IIIA Mice. JIMD Rep. 2013, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Sivakumur, P.; Wraith, J.E. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J. Inherit. Metab. Dis. 1999, 22, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Welling, L.; Marchal, J.P.; van Hasselt, P.; van der Ploeg, A.T.; Wijburg, F.A.; Boelens, J.J. Early Umbilical Cord Blood-Derived Stem Cell Transplantation Does Not Prevent Neurological Deterioration in Mucopolysaccharidosis Type III. JIMD Rep. 2015, 18, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dodsworth, C.; Paras, A.; Burton, B.K. High dose genistein aglycone therapy is safe in patients with mucopolysaccharidoses involving the central nervous system. Mol. Genet. Metab. 2013, 109, 382–385. [Google Scholar] [CrossRef]
- Heldermon, C.D.; Qin, E.Y.; Ohlemiller, K.K.; Herzog, E.D.; Brown, J.R.; Vogler, C.; Hou, W.; Orrock, J.L.; Crawford, B.E.; Sands, M.S. Disease correction by combined neonatal intracranial AAV and systemic lentiviral gene therapy in Sanfilippo Syndrome type B mice. Gene Ther. 2013, 20, 913–921. [Google Scholar] [CrossRef]
- Heldermon, C.D.; Ohlemiller, K.K.; Herzog, E.D.; Vogler, C.; Qin, E.; Wozniak, D.F.; Tan, Y.; Orrock, J.L.; Sands, M.S. Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy, or both in the mouse model of MPS IIIB. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 873–880. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Orii, T. Mucopolysaccharidosis IV (Morquio Syndrome; MPS IV). In Lysosomal Storage Disorders; Barranger, J.A., Cabrera-Salazar, M., Eds.; Springer US: Boston, MA, USA, 2007; pp. 433–445. ISBN 978-0-387-70909-3. [Google Scholar]
- Montaño, A.; Tomatsu, S.; Gottesman, G.; Smith, M.; Orii, T. International Morquio A Registry: Clinical manifestation and natural course of Morquio A disease. J. Inherit. Metab. Dis. 2007, 30, 165–174. [Google Scholar] [CrossRef]
- Harmatz, P.; Mengel, K.E.; Giugliani, R.; Valayannopoulos, V.; Lin, S.P.; Parini, R.; Guffon, N.; Burton, B.K.; Hendriksz, C.J.; Mitchell, J.; et al. The Morquio A Clinical Assessment Program: Baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol. Genet. Metab. 2013, 109, 54–61. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Villarreal, M.S.; Savarirayan, R. International guidelines for the management and treatment of Morquio A syndrome. Am. J. Med. Genet. Part A 2015, 167, 11–25. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Harmatz, P.; Beck, M.; Jones, S.; Wood, T.; Lachman, R.; Gravance, C.G.; Orii, T.; Tomatsu, S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol. Genet. Metab. 2013, 110, 54–64. [Google Scholar] [CrossRef]
- Arbisser, A.I.; Donnelly, K.A.; Scott, C.I.; DiFerrante, N.; Singh, J.; Stevenson, R.E.; Aylesworth, A.S.; Howell, R.R. Morquio-like syndrome with beta galactosidase deficiency and normal hexosamine sulfatase activity: Mucopolysacchariodosis IVB. Am. J. Med. Genet. 1977, 1, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Kecskemethy, H.H.; Harcke, H.T.; Tomatsu, S. Bone mineral density in mucopolysaccharidosis IVB. Mol. Genet. Metab. Rep. 2016, 8, 80–84. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.S.; Gugler, E.; Giedion, A.; Wiessmann, U.; Herschkowitz1, N.; Meier, C.; Leroy, J. Spondyloepiphyseal dysplasia, corneal clouding, normal intelligence and acid β-galactosidase deficiency. Clin. Genet. 1976, 9, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Santra, S.; Jones, S.A.; Geberhiwot, T.; Jesaitis, L.; Long, B.; Qi, Y.; Hawley, S.M.; Decker, C. Safety, immunogenicity, and clinical outcomes in patients with Morquio A syndrome participating in 2 sequential open-label studies of elosulfase alfa enzyme replacement therapy (MOR-002/MOR-100), representing 5 years of treatment. Mol. Genet. Metab. 2018, 123, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Mengel, E.; Guffon, N.; Valayannopoulos, V.; Parini, R.; Hughes, D.; Pastores, G.M.; Lau, H.A.; et al. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. SI Lysosome 2015, 114, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Sawamoto, K.; Almeciga-Diaz, C.J.; Shimada, T.; Bober, M.B.; Chinen, Y.; Yabe, H.; Montaño, A.M.; Giugliani, R.; Kubaski, F.; et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des. Devel. Ther. 2015, 9, 1937–1953. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Nakamura, K. Elosulfase alfa enzyme replacement therapy attenuates disease progression in a non-ambulatory Japanese patient with Morquio A syndrome (case report). Mol. Genet. Metab. Rep. 2017, 13, 76–79. [Google Scholar] [CrossRef]
- Chinen, Y.; Higa, T.; Tomatsu, S.; Suzuki, Y.; Orii, T.; Hyakuna, N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol. Genet. Metab. Rep. 2014, 1, 31–41. [Google Scholar] [CrossRef]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Tomatsu, S.; Barrera, L.A. Adeno-associated virus gene transfer in Morquio A disease—Effect of promoters and sulfatase-modifying factor 1. FEBS J. 2010, 277, 3608–3619. [Google Scholar] [CrossRef]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Barrera, L.A.; Tomatsu, S. Tailoring the AAV2 capsid vector for bone-targeting. Pediatr. Res. 2018, 84, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Tomatsu, S. Development of Substrate Degradation Enzyme Therapy for Mucopolysaccharidosis IVA Murine Model. Int. J. Mol. Sci. 2019, 20, 4139. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Walter, C.; Parkes, W.J.; Teixido, M.; Theroux, M.C.; Szymkowski, S.; Morlet, T.; Tomatsu, S. Cochlear implantation in a patient with mucopolysaccharidosis IVA. SAGE Open Med. Case Rep. 2019, 7, 2050313X19873791. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Lampe, C.; Guffon, N.; Ketteridge, D.; Leao-Teles, E.; Wraith, J.E.; Jones, S.A.; Piscia-Nichols, C.; Lin, P.; Quartel, A.; et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)--10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am. J. Med. Genet. Part A 2014, 164, 1953–1964. [Google Scholar] [CrossRef] [PubMed]
- Akyol, M.U.; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; Eto, Y.; Gold, J.I.; et al. Recommendations for the management of MPS VI: Systematic evidence-and consensus-based guidance. Orphanet J. Rare Dis. 2019, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Chintalapati, K.; Tomatsu, S.; Morlet, T.; Nagao, K. Longitudinal analysis of hearing loss in mucopolysaccharidosis types IV and VI. In Proceedings of the 10th Annual Undergraduate Research & Service Scholar Celebratory Symposium, University of Delaware, Newark, DE, USA, 15 August 2019; Available online: https://www.urp.udel.edu/summer-sym-pres/longitudinal-analysis-of-hearing-loss-in-mucopolysaccharidosis-types-iv-and-vi/ (accessed on 3 August 2020).
- Shigematsu, Y.; Hori, C.; Nakai, A.; Kuriyama, M.; Kikawa, Y.; Konishi, Y.; Sudo, M.; Konishi, K. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) with hearing impairment and pupillary membrane remnants. Acta Paediatrics Int. 1991, 33, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Pellico, A.; Pittalà, A.; Gasperini, S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 121. [Google Scholar] [CrossRef]
- Guffon, N.; Froissart, R.; Fouilhoux, A. A rare late progression form of Sly syndrome mucopolysaccharidosis. JIMD Rep. 2019, 49, 1–6. [Google Scholar] [CrossRef]
- Sands, M.S.; Vogler, C.; Torrey, A.; Levy, B.; Gwynn, B.; Grubb, J.; Sly, W.S.; Birkenmeier, E.H. Murine mucopolysaccharidosis type VII: Long term therapeutic effects of enzyme replacement and enzyme replacement followed by bone marrow transplantation. J. Clin. Investig. 1997, 99, 1596–1605. [Google Scholar] [CrossRef][Green Version]
- Sands, M.S.; Birkenmeier, E.H. A single-base-pair deletion in the beta-glucuronidase gene accounts for the phenotype of murine mucopolysaccharidosis type VII. Proc. Natl. Acad. Sci. USA 1993, 90, 6567–6571. [Google Scholar] [CrossRef]
- Cadaoas, J.; Boyle, G.; Jungles, S.; Cullen, S.; Vellard, M.; Grubb, J.H.; Jurecka, A.; Sly, W.; Kakkis, E. Vestronidase alfa: Recombinant human β-glucuronidase as an enzyme replacement therapy for MPS VII. Mol. Genet. Metab. 2020, 130, 65–76. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2019, 33, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.C.; Atmuri, V.; Hemming, R.J.; Farley, J.; Mort, J.S.; Byers, S.; Hombach-Klonisch, S.; Csoka, A.B.; Stern, R.; Triggs-Raine, B.L. A mouse model of human mucopolysaccharidosis IX exhibits osteoarthritis. Hum. Mol. Genet. 2008, 17, 1904–1915. [Google Scholar] [CrossRef]
- Triggs-Raine, B.; Natowicz, M.R. Biology of hyaluronan: Insights from genetic disorders of hyaluronan metabolism. World J. Biol. Chem. 2015, 6, 110–120. [Google Scholar] [CrossRef]
- Jadin, L.; Wu, X.; Ding, H.; Frost, G.I.; Onclinx, C.; Triggs-Raine, B.; Flamion, B. Skeletal and hematological anomalies in HYAL2-deficient mice: A second type of mucopolysaccharidosis IX? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 4316–4326. [Google Scholar] [CrossRef]
- Węgrzyn, G.; Jakóbkiewicz-Banecka, J.; Narajczyk, M.; Wiśniewski, A.; Piotrowska, E.; Gabig-Cimińska, M.; Kloska, A.; Słomińska-Wojewódzka, M.; Korzon-Burakowska, A.; Węgrzyn, A. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Med. Hypotheses 2010, 75, 605–609. [Google Scholar] [CrossRef]
- Bianchi, P.M.; Gaini, R.; Vitale, S. ENT and mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 127. [Google Scholar] [CrossRef]
- Torres, D.A.; Barth, A.L.; Valente, M.P.M.; Mello, P.P.D.; Horovitz, D.D.G. Otolaryngologists and the Early Diagnosis of Mucopolysaccharidoses: A Cross-Sectional Study. Diagn. Basel Switz. 2019, 9, 187. [Google Scholar] [CrossRef]
- Wold, S.M.; Derkay, C.S.; Darrow, D.H.; Proud, V. Role of the pediatric otolaryngologist in diagnosis and management of children with mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 27–31. [Google Scholar] [CrossRef]


{kind=link}
| MPS Subtype | Enzyme | GAGs |
|---|---|---|
| MPS I (Hurler syndrome) | alpha-L-iduronidase [1] | DS and HS [1] |
| MPS II (Hunter syndrome) | iduronate-2-sulfatase [2] | DS and HS [1] |
| MPS IIIA (Sanfilippo syndrome type A) | heparan N-sulfatase [3] | HS [1] |
| MPS IIIB (Sanfilippo syndrome type B) | alpha-N-acetylglucosaminidase [3] | HS [1] |
| MPS IIIC (Sanfilippo syndrome type C) | acetyl CoA alpha-glucosaminide acetyltransferase [3] | HS [1] |
| MPS IIID (Sanfilippo syndrome type D) | N-acetylglucosamine 6-sulfatase [3] | HS [1] |
| MPS IVA (Morquio syndrome type A) | N-acetylgalactosamine-6-sulfate sulfatase [4] | Chondroitin-6-sulfate and KS [4] |
| MPS IV B (Morquio syndrome type B) | beta-galactosidase [5] | KS [5] |
| MPS VI (Maroteaux–Lamy syndrome) | arylsulfatase B [6] | DS [1] |
| MPS VII (Sly syndrome) | glucuronidase [1] | Chondroitin sulfate, DS, and HS [1] |
| MPS IX (Natowicz syndrome) | hyaluronidase [7] | Hyaluronic acid [7] |
| Source | MPS Type(s) | Article Type (Research, Case Study, Review) | N | Animal/Human |
|---|---|---|---|---|
| Simmons et al., 2005 [9] | All MPS types | Retrospective review | N/A | Human |
| Silveira et al., 2018 [10] | I, II, III, IV, VI | Descriptive, cross-sectional study | 53 | Human |
| Lenka et al., 2020 [11] | I, II, III, IV, VI | Retrospective review | 61 | Human |
| Ahn et al., 2019 [12] | I, II, III, IV, VI | Retrospective review | 124 | Human |
| Mesolella et al., 2013 [13] | I, II, III, IV, VI | Observational Study | 20 | Human |
| Lin et al., 2014 [14] | I, II, IV, VI | Clinical study | 39 | Human |
| Vargas-Gamarra et al., 2017 [15] | I, II, III, IV | Retrospective study | 23 | Human |
| Gokdogan et al., 2016 [16] | I, III, IV, VI | Clinical study | 9 | Human |
| Giraldo et al., 2020 [17] | II, IVA, VI | Retrospective study | 35 | Human |
| Da Costa et al., 2012 [18] | I, II | Retrospective study | 30 | Human |
| van den Broek et al., 2020 [19] | I, VI | Retrospective study | 32 | Human |
| Aldenhoven et al., 2015 [20] | I | Retrospective study | 217 | Human |
| Dualibi et al., 2016 [21] | I | Prospective study | 9 | Human |
| Kiely et al., 2017 [22] | I | Retrospective review | 55 | Human |
| Friedmann et al., 1985 [23] | I | Histopathological study | 2 | Human |
| Kariya et al., 2012 [24] | I | Temporal bone scan study | 6 | Human |
| Souillet et al., 2003 [25] | I | Prospective study | 27 | Human |
| Schachern et al., 2007 [26] | I | Research study | N/A | Mouse |
| Schachern et al., 1984 [27] | I | Temporal bone scan study | 3 | Human |
| Keilmann et al., 2012 [28] | II | Survey/Registry | 554 | Human |
| Chiong et al., 2017 [29] | II | Case series | 23 | Human |
| Muenzer et al., 2006 [30] | II | Clinical trial | 96 | Human |
| Hong et al., 2012 [31] | II | Research study | N/A | Mouse |
| Buhrman et al. 2014 [32] | IIIA | Retrospective review | 46 | Human |
| Heldermon et al., 2007 [33] | IIIB | Research study | N/A | Mouse |
| Nagao et al., 2018 [34] | IVA, IVB | Clinical study | 14 | Human |
| Riedner and Levin, 1977 [35] | IV | Audiological/Otologic review | 21 | Human |
| Swartz and Santi, 1997 [36] | IV | Animal research | N/A | Animal (chinchilla, cat, gerbil, rabbit) |
| Gomes et al., 2019 [37] | VI | Clinical review | 362 | Human |
| Furujo et al., 2017 [38] | VI | Case study | 2 | Human |
| Harmatz et al., 2014 [39] | VI | Clinical trial review | N/A | Human |
| Horovitz et al., 2013 [40] | VI | Retrospective review | 34 | Human |
| Montaño et al., 2016 [41] | VII | Survey | 56 | Human |
| Ohlemiller et al., 2002 [42] | VII | Research study | N/A | Mouse |
| O’Connor et al., 1998 [43] | VII | Research study | N/A | Mouse |
| Sands et al., 1995 [44] | VII | Research study | N/A | Mouse |
| Berry et al., 1994 [45] | VII | Research study | N/A | Mouse |
| Natowicz et al., 1996 [7] | IX | Case report | 1 | Human |
| Imundo et al., 2011 [46] | IX | Clinical case reports | 3 | Human |
| Type | Enzyme | OMIM Number | Gene |
|---|---|---|---|
| MPS IIIA | Heparan N-sulfatase | 252900 | SGSH |
| MPS IIIB | Alpha-N-acetylglucosaminidase | 252920 | NAGLU |
| MPS IIIC | Acetyl CoA alpha-glucosaminide acetyltransferase | 252930 | HGSNAT |
| MPS IIID | N-acetylglucosamine 6-sulfatase | 252940 | GNS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfberg, J.; Chintalapati, K.; Tomatsu, S.; Nagao, K. Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions. Diagnostics 2020, 10, 554. https://doi.org/10.3390/diagnostics10080554
Wolfberg J, Chintalapati K, Tomatsu S, Nagao K. Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions. Diagnostics. 2020; 10(8):554. https://doi.org/10.3390/diagnostics10080554
Chicago/Turabian StyleWolfberg, Jeremy, Keerthana Chintalapati, Shunji Tomatsu, and Kyoko Nagao. 2020. "Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions" Diagnostics 10, no. 8: 554. https://doi.org/10.3390/diagnostics10080554
APA StyleWolfberg, J., Chintalapati, K., Tomatsu, S., & Nagao, K. (2020). Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions. Diagnostics, 10(8), 554. https://doi.org/10.3390/diagnostics10080554