Revisiting Cell Death Responses in Fibrotic Lung Disease: Crosstalk between Structured and Non-Structured Cells

Abstract
1. Introduction
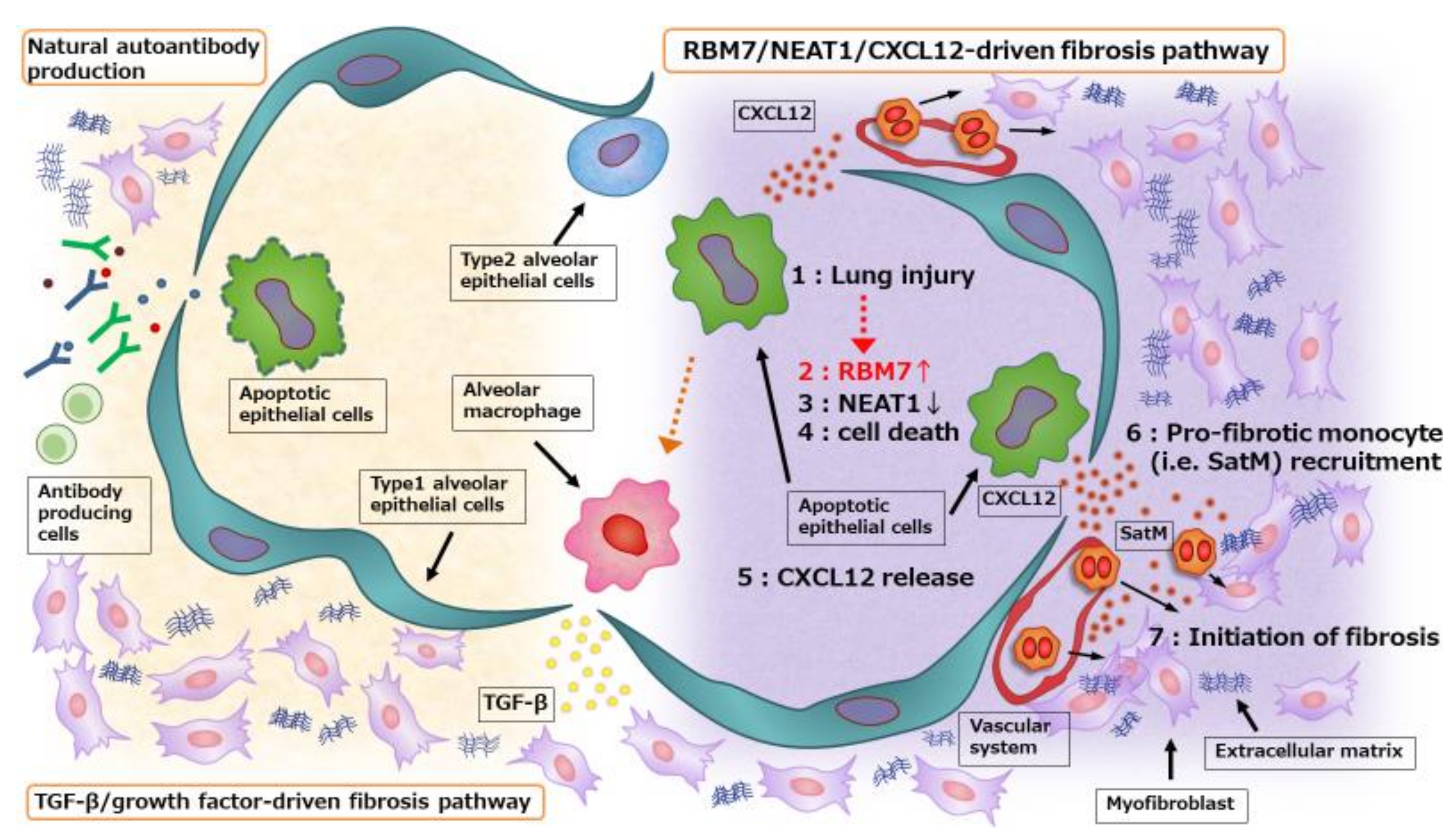
2. Cell Death in Structured Cells Initiates Fibrosis Development
3. Interaction between Structured and Non-Structured Cells in the Development of Fibrosis
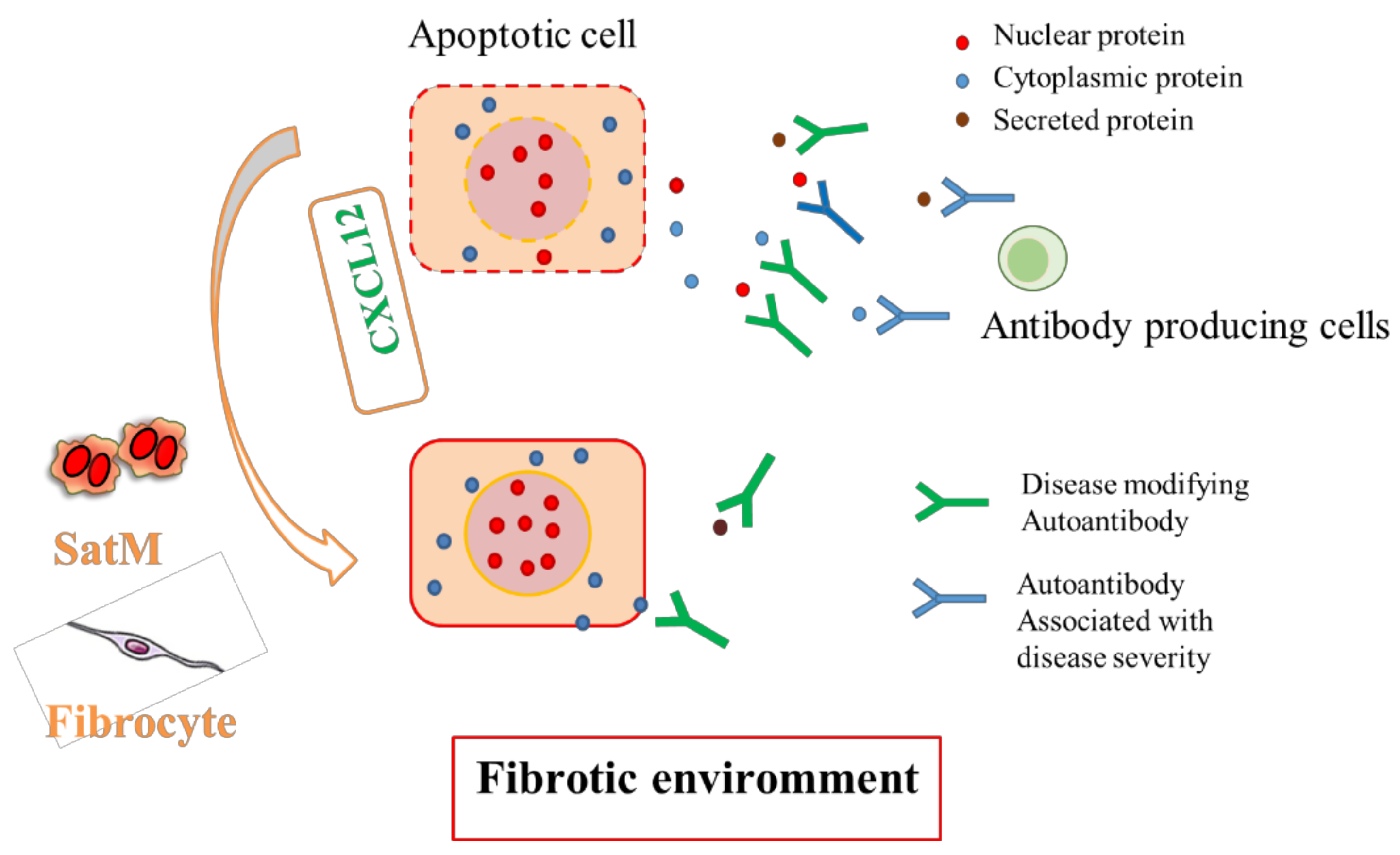
4. Natural Autoantibodies in Fibrotic Lung Disease
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Hoo, Z.H.; Whyte, M.K. Idiopathic pulmonary fibrosis. Thorax 2012, 67, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic pulmonary fibrosis: From epithelial injury to biomarkers--insights from the bench side. Respir. Int. Rev. Thorac. Dis. 2013, 86, 441–452. [Google Scholar] [CrossRef]
- Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. New Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef]
- Fukushima, K.; Satoh, T.; Sugihara, F.; Sato, Y.; Okamoto, T.; Mitsui, Y.; Yoshio, S.; Li, S.; Nojima, S.; Motooka, D.; et al. Dysregulated Expression of the Nuclear Exosome Targeting Complex Component Rbm7 in Nonhematopoietic Cells Licenses the Development of Fibrosis. Immunity 2020, 52, 542–556. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef]
- Palma, J.; Tokarz-Deptula, B.; Deptula, J.; Deptula, W. Natural antibodies–facts known and unknown. Cent. Eur. J. Immunol. 2018, 43, 466–475. [Google Scholar] [CrossRef]
- Nagele, E.P.; Han, M.; Acharya, N.K.; DeMarshall, C.; Kosciuk, M.C.; Nagele, R.G. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE 2013, 8, e60726. [Google Scholar] [CrossRef]
- Fukushima, K.; Tsujino, K.; Futami, S.; Kida, H. Natural Autoantibodies in Chronic Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 1138. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Kishimoto, T.; Ashizawa, K.; Kato, K.; Okamoto, K.; Honma, K.; Hayashi, S.; Akira, M. Asbestosis and other pulmonary fibrosis in asbestos-exposed workers: High-resolution CT features with pathological correlations. Eur. Radiol. 2016, 26, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 2011, 17, 355–361. [Google Scholar] [CrossRef]
- Mishra, A.; Doyle, N.A.; Martin, W.J.C. Bleomycin-mediated pulmonary toxicity: Evidence for a p53-mediated response. Am. J. Respir. Cell Mol. Biol. 2000, 22, 543–549. [Google Scholar] [CrossRef]
- Hagimoto, N.; Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hara, N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997, 16, 91–101. [Google Scholar] [CrossRef]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Maeyama, T.; Hagimoto, N.; Kawasaki, M.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L316–L325. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ibarra-Sunga, O.; Verlinski, L.; Pick, R.; Uhal, B.D. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L143–L151. [Google Scholar] [CrossRef] [PubMed]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Schuster, N.; Krieglstein, K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002, 307, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- Lubas, M.; Christensen, M.S.; Kristiansen, M.S.; Domanski, M.; Falkenby, L.G.; Lykke-Andersen, S.; Andersen, J.S.; Dziembowski, A.; Jensen, T.H. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell 2011, 43, 624–637. [Google Scholar] [CrossRef]
- Sofos, N.; Winkler, M.B.; Brodersen, D.E. RRM domain of human RBM7: Purification, crystallization and structure determination. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 397–402. [Google Scholar] [CrossRef]
- Tiedje, C.; Lubas, M.; Tehrani, M.; Menon, M.B.; Ronkina, N.; Rousseau, S.; Cohen, P.; Kotlyarov, A.; Gaestel, M. p38MAPK/MK2-mediated phosphorylation of RBM7 regulates the human nuclear exosome targeting complex. RNA 2015, 21, 262–278. [Google Scholar] [CrossRef]
- Giunta, M.; Edvardson, S.; Xu, Y.; Schuelke, M.; Gomez-Duran, A.; Boczonadi, V.; Elpeleg, O.; Muller, J.S.; Horvath, R. Altered RNA metabolism due to a homozygous RBM7 mutation in a patient with spinal motor neuropathy. Hum. Mol. Genet. 2016, 25, 2985–2996. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Naganuma, T.; Hirose, T. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 2013, 10, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Lamond, A.I. Paraspeckles. Cold Spring Harb. Perspect. Biol. 2010, 2, a000687. [Google Scholar] [CrossRef] [PubMed]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.; Radaelli, E.; Vermi, W.; et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Crystal, R.G.; Bitterman, P.B.; Mossman, B.; Schwarz, M.I.; Sheppard, D.; Almasy, L.; Chapman, H.A.; Friedman, S.L.; King, T.E., Jr.; Leinwand, L.A.; et al. Future research directions in idiopathic pulmonary fibrosis: Summary of a National Heart, Lung, and Blood Institute working group. Am. J. Respir. Crit. Care Med. 2002, 166, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Dunay, I.R.; Fuchs, A.; Sibley, L.D. Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect. Immun. 2010, 78, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Nenan, S.; Guenon, I.; Lagente, V.; Boichot, E. Influence of early neutrophil depletion on MMPs/TIMP-1 balance in bleomycin-induced lung fibrosis. Int. Immunopharmacol. 2007, 7, 900–911. [Google Scholar] [CrossRef]
- Aubin Vega, M.; Chupin, C.; Pascariu, M.; Prive, A.; Dagenais, A.; Berthiaume, Y.; Brochiero, E. Dexamethasone fails to improve bleomycin-induced acute lung injury in mice. Physiol. Rep. 2019, 7, e14253. [Google Scholar] [CrossRef]
- Kim, K.K.; Dotson, M.R.; Agarwal, M.; Yang, J.; Bradley, P.B.; Subbotina, N.; Osterholzer, J.J.; Sisson, T.H. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018, 9, 1056. [Google Scholar] [CrossRef]
- De Oliveira Fulco, T.; Andrade, P.R.; de Mattos Barbosa, M.G.; Pinto, T.G.; Ferreira, P.F.; Ferreira, H.; da Costa Nery, J.A.; Real, S.C.; Borges, V.M.; Moraes, M.O.; et al. Effect of apoptotic cell recognition on macrophage polarization and mycobacterial persistence. Infect. Immun. 2014, 82, 3968–3978. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Frasch, S.C.; Thomas, S.M.; Bratton, D.L.; Henson, P.M. Induction of TGF-beta1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor CD36. PLoS ONE 2013, 8, e72772. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Wu, S.B.; Kau, H.C.; Wei, Y.H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-beta1 (TGF-beta1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef]
- Peng, H.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Curr. Opin. Pharmacol. 2012, 12, 491–496. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef]
- Nagasawa, T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. 2014, 92, 433–439. [Google Scholar] [CrossRef]
- Tashiro, K.; Tada, H.; Heilker, R.; Shirozu, M.; Nakano, T.; Honjo, T. Signal sequence trap: A cloning strategy for secreted proteins and type I membrane proteins. Science 1993, 261, 600–603. [Google Scholar] [CrossRef]
- Nagasawa, T.; Kikutani, H.; Kishimoto, T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc. Natl. Acad. Sci. USA 1994, 91, 2305–2309. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Yellowley, C.E.; Toupadakis, C.A.; Vapniarsky, N.; Wong, A. Circulating progenitor cells and the expression of Cxcl12, Cxcr4 and angiopoietin-like 4 during wound healing in the murine ear. PLoS ONE 2019, 14, e0222462. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, X.; Langer, M.; Weber, C.; Koenen, R.R.; von Hundelshausen, P. Touch of chemokines. Front. Immunol. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Rose, J.J.; Lodge, R.; Murphy, P.M.; Foley, J.F. Distinct mechanisms of agonist-induced endocytosis for human chemokine receptors CCR5 and CXCR4. Mol. Biol. Cell 2003, 14, 3305–3324. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Benovic, J.L. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 2001, 276, 45509–45512. [Google Scholar] [CrossRef]
- Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D.; Dubrovska, A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347–1361. [Google Scholar] [CrossRef]
- Dorman, S.C.; Babirad, I.; Post, J.; Watson, R.M.; Foley, R.; Jones, G.L.; O’Byrne, P.M.; Sehmi, R. Progenitor egress from the bone marrow after allergen challenge: Role of stromal cell-derived factor 1alpha and eotaxin. J. Allergy Clin. Immunol. 2005, 115, 501–507. [Google Scholar] [CrossRef]
- Hoshino, M.; Aoike, N.; Takahashi, M.; Nakamura, Y.; Nakagawa, T. Increased immunoreactivity of stromal cell-derived factor-1 and angiogenesis in asthma. Eur. Respir. J. 2003, 21, 804–809. [Google Scholar] [CrossRef]
- Petty, J.M.; Sueblinvong, V.; Lenox, C.C.; Jones, C.C.; Cosgrove, G.P.; Cool, C.D.; Rai, P.R.; Brown, K.K.; Weiss, D.J.; Poynter, M.E.; et al. Pulmonary stromal-derived factor-1 expression and effect on neutrophil recruitment during acute lung injury. J. Immunol. 2007, 178, 8148–8157. [Google Scholar] [CrossRef]
- Xu, J.; Mora, A.; Shim, H.; Stecenko, A.; Brigham, K.L.; Rojas, M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Jin, H.; Liu, T.; Chensue, S.W.; Phan, S.H. Bone marrow-derived progenitor cells in pulmonary fibrosis. J. Clin. Investig. 2004, 113, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.N.; Schreiner, P.; Ng, B.Y.; Lo, B.; Hughes, M.R.; Scott, R.W.; Gusti, V.; Lecour, S.; Simonson, E.; Manisali, I.; et al. Impact of a CXCL12/CXCR4 Antagonist in Bleomycin (BLM) Induced Pulmonary Fibrosis and Carbon Tetrachloride (CCl4) Induced Hepatic Fibrosis in Mice. PLoS ONE 2016, 11, e0151765. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Aono, Y.; Azuma, M.; Kishi, M.; Yokota, Y.; Kinoshita, K.; Takezaki, A.; Kishi, J.; Kawano, H.; Ogawa, H.; et al. Antifibrotic effects of CXCR4 antagonist in bleomycin-induced pulmonary fibrosis in mice. J. Med. Investig. JMI 2013, 60, 127–137. [Google Scholar] [CrossRef]
- Costantini, S.; Raucci, R.; Colonna, G.; Mercurio, F.A.; Trotta, A.M.; Paola, R.; Leone, M.; Rossi, F.; Pellegrino, C.; Castello, G.; et al. Peptides targeting chemokine receptor CXCR4: Structural behavior and biological binding studies. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2014, 20, 270–278. [Google Scholar] [CrossRef]
- Zhang, W.B.; Navenot, J.M.; Haribabu, B.; Tamamura, H.; Hiramatu, K.; Omagari, A.; Pei, G.; Manfredi, J.P.; Fujii, N.; Broach, J.R.; et al. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J. Biol. Chem. 2002, 277, 24515–24521. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, F.; Wang, X.; Yao, W.; Wang, M.; Pei, G.; Hu, Z.; Guo, Y.; Zhao, Z.; Wang, P.; et al. Continuous AMD3100 Treatment Worsens Renal Fibrosis through Regulation of Bone Marrow Derived Pro-Angiogenic Cells Homing and T-Cell-Related Inflammation. PLoS ONE 2016, 11, e0149926. [Google Scholar] [CrossRef]
- Saiman, Y.; Jiao, J.; Fiel, M.I.; Friedman, S.L.; Aloman, C.; Bansal, M.B. Inhibition of the CXCL12/CXCR4 chemokine axis with AMD3100, a CXCR4 small molecule inhibitor, worsens murine hepatic injury. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 794–803. [Google Scholar] [CrossRef]
- Abraham, M.; Pereg, Y.; Bulvik, B.; Klein, S.; Mishalian, I.; Wald, H.; Eizenberg, O.; Beider, K.; Nagler, A.; Golan, R.; et al. Single Dose of the CXCR4 Antagonist BL-8040 Induces Rapid Mobilization for the Collection of Human CD34(+) Cells in Healthy Volunteers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6790–6801. [Google Scholar] [CrossRef]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Gough, W.; Stewart, J.; Uhlik, M.T.; Chen, Q.; Hui, Y.H.; Zamek-Gliszczynski, M.J.; et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol. Cancer Ther. 2015, 14, 480–490. [Google Scholar] [CrossRef]
- Gabasa, M.; Ikemori, R.; Hilberg, F.; Reguart, N.; Alcaraz, J. Nintedanib selectively inhibits the activation and tumour-promoting effects of fibroblasts from lung adenocarcinoma patients. Br. J. Cancer 2017, 117, 1128–1138. [Google Scholar] [CrossRef]
- Lin, C.H.; Shih, C.H.; Tseng, C.C.; Yu, C.C.; Tsai, Y.J.; Bien, M.Y.; Chen, B.C. CXCL12 induces connective tissue growth factor expression in human lung fibroblasts through the Rac1/ERK, JNK, and AP-1 pathways. PLoS ONE 2014, 9, e104746. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Nemtsov, A.; Neufeld, M.; Rehm, J. Are Trends in Alcohol Consumption and Cause-Specific Mortality in Russia Between 1990 and 2017 the Result of Alcohol Policy Measures? J. Stud. Alcohol Drugs 2019, 80, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.; Hughes, J. Apoptosis and autoimmunity. Nephrol. Dial. Transplant. 2002, 17, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Park, J.K.; Roh, J.H.; Song, J.W.; Lee, C.K.; Kim, M.; Jang, S.J.; Colby, T.V.; Kim, D.S. Clinical significance of serum autoantibodies in idiopathic interstitial pneumonia. J. Korean Med. Sci. 2013, 28, 731–737. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.A.; Wilkes, D.S. Autoimmunity in idiopathic pulmonary fibrosis: Are circulating autoantibodies pathogenic or epiphenomena? Am. J. Respir. Crit. Care Med. 2011, 183, 692–693. [Google Scholar] [CrossRef]
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Watanabe, K.; Aihara, K.; Ikezoe, K.; Sokai, A.; Nakatsuka, Y.; et al. The long-term outcome of interstitial lung disease with anti-aminoacyl-tRNA synthetase antibodies. Respir. Med. 2017, 127, 57–64. [Google Scholar] [CrossRef]
- Sato, S.; Hoshino, K.; Satoh, T.; Fujita, T.; Kawakami, Y.; Kuwana, M. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009, 60, 2193–2200. [Google Scholar] [CrossRef]
- Yoshifuji, H.; Fujii, T.; Kobayashi, S.; Imura, Y.; Fujita, Y.; Kawabata, D.; Usui, T.; Tanaka, M.; Nagai, S.; Umehara, H.; et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006, 39, 233–241. [Google Scholar] [CrossRef]
- Kochi, Y.; Kamatani, Y.; Kondo, Y.; Suzuki, A.; Kawakami, E.; Hiwa, R.; Momozawa, Y.; Fujimoto, M.; Jinnin, M.; Tanaka, Y.; et al. Splicing variant of WDFY4 augments MDA5 signalling and the risk of clinically amyopathic dermatomyositis. Ann. Rheum. Dis. 2018, 77, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Hamano, Y.; Kida, H.; Ihara, S.; Murakami, A.; Yanagawa, M.; Ueda, K.; Honda, O.; Tripathi, L.P.; Arai, T.; Hirose, M.; et al. Classification of idiopathic interstitial pneumonias using anti-myxovirus resistance-protein 1 autoantibody. Sci. Rep. 2017, 7, 43201. [Google Scholar] [CrossRef] [PubMed]
- Han, M.Z.; Xu, R.; Xu, Y.Y.; Zhang, X.; Ni, S.L.; Huang, B.; Chen, A.J.; Wei, Y.Z.; Wang, S.; Li, W.J.; et al. TAGLN2 is a candidate prognostic biomarker promoting tumorigenesis in human gliomas. J. Exp. Clin. Cancer Res. CR 2017, 36, 155. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Konigshoff, M.; Jayachandran, A.; Handley, D.; Seeger, W.; Kaminski, N.; Eickelberg, O. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 1778–1789. [Google Scholar] [CrossRef]
- Gu, W.; Monteiro, R.; Zuo, J.; Simoes, F.C.; Martella, A.; Andrieu-Soler, C.; Grosveld, F.; Sauka-Spengler, T.; Patient, R. A novel TGFbeta modulator that uncouples R-Smad/I-Smad-mediated negative feedback from R-Smad/ligand-driven positive feedback. PLoS Biol. 2015, 13, e1002051. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, R.; Fang, L.; Ge, X.; Chen, L.; Zhou, M.; Zhou, Y.; Xiong, W.; Hu, Y.; Tang, X.; et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics 2019, 9, 2460–2474. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Nigdelioglu, R.; Meliton, A.Y.; Tian, Y.; Witt, L.J.; O’Leary, E.; Sun, K.A.; Woods, P.S.; Wu, D.; Ansbro, B.; et al. Inhibition of Phosphoglycerate Dehydrogenase Attenuates Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2018, 58, 585–593. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Qu, X.; Jiang, M.; Sun, Y.B.; Jiang, X.; Fu, P.; Ren, Y.; Wang, D.; Dai, L.; Caruana, G.; Bertram, J.F.; et al. The Smad3/Smad4/CDK9 complex promotes renal fibrosis in mice with unilateral ureteral obstruction. Kidney Int. 2015, 88, 1323–1335. [Google Scholar] [CrossRef]
- Yanagida, A.; Iwaisako, K.; Hatano, E.; Taura, K.; Sato, F.; Narita, M.; Nagata, H.; Asechi, H.; Uemoto, S.; Kinoshita, M. Downregulation of the Wnt antagonist Dkk2 links the loss of Sept4 and myofibroblastic transformation of hepatic stellate cells. Biochim. Biophys. Acta 2011, 1812, 1403–1411. [Google Scholar] [CrossRef][Green Version]
- Guo, F.; Jiao, D.; Sui, G.Q.; Sun, L.N.; Gao, Y.J.; Fu, Q.F.; Jin, C.X. Anticancer effect of YWHAZ silencing via inducing apoptosis and autophagy in gastric cancer cells. Neoplasma 2018, 65, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Kodama, S.; Nakano, Y.; Minaki-Nakagawa, Y.; Aoyama, Y.; Sakikubo, M.; Goto, T.; Yoshida, M.; Masui, T.; Yamamoto, T.; et al. Exocrine tissue-driven TFF2 prevents apoptotic cell death of endocrine lineage during pancreas organogenesis. Sci. Rep. 2019, 9, 1636. [Google Scholar] [CrossRef]
- He, H.; Dai, J.; Zhuo, R.; Zhao, J.; Wang, H.; Sun, F.; Zhu, Y.; Xu, D. Study on the mechanism behind lncRNA MEG3 affecting clear cell renal cell carcinoma by regulating miR-7/RASL11B signaling. J. Cell. Physiol. 2018, 233, 9503–9515. [Google Scholar] [CrossRef] [PubMed]
- Ishiguchi, H.; Izumi, H.; Torigoe, T.; Yoshida, Y.; Kubota, H.; Tsuji, S.; Kohno, K. ZNF143 activates gene expression in response to DNA damage and binds to cisplatin-modified DNA. Int. J. Cancer 2004, 111, 900–909. [Google Scholar] [CrossRef]
- Kim, Y.C.; Gonzalez-Nieves, R.; Cutler, M.L. Rsu1 contributes to cell adhesion and spreading in MCF10A cells via effects on P38 map kinase signaling. Cell Adhes. Migr. 2015, 9, 227–232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lucas, F.; Larkin, K.; Gregory, C.T.; Orwick, S.; Doong, T.J.; Lozanski, A.; Lozanski, G.; Misra, S.; Ngankeu, A.; Ozer, H.G.; et al. Novel BCL2 mutations in venetoclax-resistant, ibrutinib-resistant CLL patients with BTK/PLCG2 mutations. Blood 2020. [Google Scholar] [CrossRef]
- Huang, W.C.; Tung, S.L.; Chen, Y.L.; Chen, P.M.; Chu, P.Y. IFI44L is a novel tumor suppressor in human hepatocellular carcinoma affecting cancer stemness, metastasis, and drug resistance via regulating met/Src signaling pathway. BMC Cancer 2018, 18, 609. [Google Scholar] [CrossRef]
- Zhang, J.; Pi, J.; Liu, Y.; Yu, J.; Feng, T. Knockdown of YTH N6-methyladenosine RNA binding protein 2 (YTHDF2) inhibits proliferation and promotes apoptosis in MGC-803 gastric cancer cells. Xi Bao Yu Fen Zi Mian Yi Xue za Zhi 2017, 33, 1628–1634. [Google Scholar]
- Mojallal, M.; Zheng, Y.; Hultin, S.; Audebert, S.; van Harn, T.; Johnsson, P.; Lenander, C.; Fritz, N.; Mieth, C.; Corcoran, M.; et al. AmotL2 disrupts apical-basal cell polarity and promotes tumour invasion. Nat. Commun. 2014, 5, 4557. [Google Scholar] [CrossRef]
- Chen, Y.F.; Cho, J.J.; Huang, T.H.; Tseng, C.N.; Huang, E.Y.; Cho, C.L. Downregulation of a novel human gene, ROGDI, increases radiosensitivity in cervical cancer cells. Cancer Biol. Ther. 2016, 17, 1070–1078. [Google Scholar] [CrossRef][Green Version]
- Zheng, J.; Liu, H.; Zhu, L.; Chen, Y.; Zhao, H.; Zhang, W.; Li, F.; Xie, L.; Yan, X.; Zhu, X. Microtubule-bundling protein Spef1 enables mammalian ciliary central apparatus formation. J. Mol. Cell Biol. 2019, 11, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Blanchon, S.; Legendre, M.; Copin, B.; Duquesnoy, P.; Montantin, G.; Kott, E.; Dastot, F.; Jeanson, L.; Cachanado, M.; Rousseau, A.; et al. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J. Med. Genet. 2012, 49, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Collawn, J.F.; Fu, L.; Bebok, Z. Targets for cystic fibrosis therapy: Proteomic analysis and correction of mutant cystic fibrosis transmembrane conductance regulator. Expert Rev. Proteom. 2010, 7, 495–506. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Signalling Pathways | Antigens for IPF Specific Natural AutoAbs |
|---|---|
| TGF-β and fibroblast activation | transgelin 2 [83], transgelin 3 [84], LIM domain-binding protein 2 [85], HLA complex P5 [86], PHGDH [87], NAT6 [88], CDK9 [89], SEPT4 [90] |
| cell death regulation | 14-3-3 protein zeta/delta [91], Trefoil factor 2 protein [92], RAS-like family 11 member B [93], MRPS11 [94], RSU1 [95], PLCG2 [96], IFI44L [97], YTHDF2 [98], AMOTL2 [99], ROGDI [100] |
| airway clearance | sperm flagellar 1 [101], cilia and flagella associated protein 410 [102], t-complex 10 like [103] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukushima, K.; Satoh, T.; Kida, H.; Kumanogoh, A. Revisiting Cell Death Responses in Fibrotic Lung Disease: Crosstalk between Structured and Non-Structured Cells. Diagnostics 2020, 10, 504. https://doi.org/10.3390/diagnostics10070504
Fukushima K, Satoh T, Kida H, Kumanogoh A. Revisiting Cell Death Responses in Fibrotic Lung Disease: Crosstalk between Structured and Non-Structured Cells. Diagnostics. 2020; 10(7):504. https://doi.org/10.3390/diagnostics10070504
Chicago/Turabian StyleFukushima, Kiyoharu, Takashi Satoh, Hiroshi Kida, and Atsushi Kumanogoh. 2020. "Revisiting Cell Death Responses in Fibrotic Lung Disease: Crosstalk between Structured and Non-Structured Cells" Diagnostics 10, no. 7: 504. https://doi.org/10.3390/diagnostics10070504
APA StyleFukushima, K., Satoh, T., Kida, H., & Kumanogoh, A. (2020). Revisiting Cell Death Responses in Fibrotic Lung Disease: Crosstalk between Structured and Non-Structured Cells. Diagnostics, 10(7), 504. https://doi.org/10.3390/diagnostics10070504