Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy


 and
and
Abstract
1. Introduction
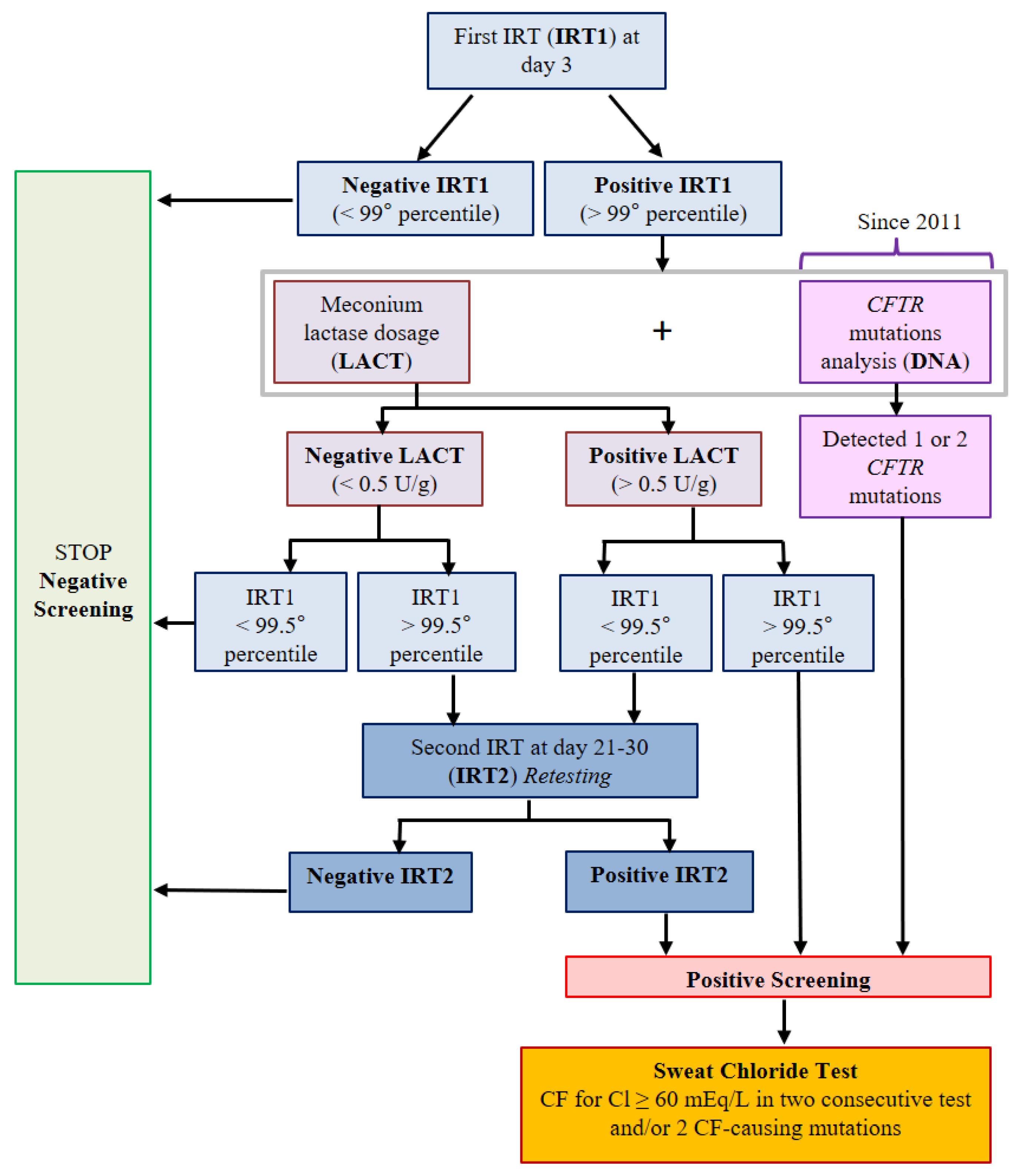
2. Materials and Methods
- Respiratory (bronchopulmonary infections, chronic cough, expectoration)
- Gastroenteric (irregular defecation, frequent episodes of diarrhea)
- Failure to thrive (not meeting standards of growth)
- Salt-loss syndrome (dehydration with metabolic hypochloremic alkalosis)
- Nasal polyposis
- Pancreatitis
- Liver disease
- Diabetes
- Distal bowel obstruction syndrome (DIOS)
- Azoospermia
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ratjen, F.; Doring, G. Cystic fibrosis. Lancet 2003, 361, 681–689. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Tsui, L.; Buchwald, M.; Barker, D.; Braman, J.; Knowlton, R.; Schumm, J.; Eiberg, H.; Mohr, J.; Kennedy, D.; Plavsic, N.; et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985, 230, 1054–1057. [Google Scholar] [CrossRef]
- Morral, N.; Bertranpetit, J.; Estivill, X.; Nunes, V.; Casals, T.; Giménez, J.; Reis, A.; Varon-Mateeva, R.; Macek, M.; Kalaydjieva, L.; et al. The origin of the major cystic fibrosis mutation (ΔF508) in European populations. Nat. Genet. 1994, 7, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Tosco, A.; Tomaiuolo, R.; Sepe, A.; Amato, N.; Casale, A.; Mercogliano, C.; De Gregorio, F.; Improta, F.; Elce, A.; et al. Prediction of acute pancreatitis risk based on PIP score in children with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 579–584. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Terlizzi, V.; Lucarelli, M.; Salvatore, D.; Angioni, A.; Bisogno, A.; Braggion, C.; Buzzetti, R.; Carnovale, V.; Casciaro, R.; Castaldo, G.; et al. Clinical expression of cystic fibrosis in a large cohort of Italian siblings. BMC Pulm. Med. 2018, 18, 196. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Patient Registry Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2016. [Google Scholar]
- Castellani, C.; Massie, J.; Sontag, M.; Southern, K.W. Newborn screening for cystic fibrosis. Lancet Respir. Med. 2016, 4, 653–661. [Google Scholar] [CrossRef]
- Pederzini, F.; Cabrini, G.; Faraguna, D.; Giglio, L.; Mengarda, G.; Pedrotti, D.; Mastella, G. Neonatal screening for cystic fibrosis using blood trypsin with complementary meconium lactase: An advisable strategy for the population of southern Europe. Screening 1995, 3, 173–179. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.; Bell, S.C.; Heijerman, H.G.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef] [PubMed]
- Barben, J.; Castellani, C.; Dankert-Roelse, J.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.; Munck, A.; Sands, D.; Sommerburg, O.; et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J. Cyst. Fibros. 2016, 16, 207–213. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2014 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2015. [Google Scholar]
- Giordani, B.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Minicucci, L.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; et al. Last Italian Register of Cystic Fibrosis. Report 2015–2016. Epidemiol. Prev. 2019, 43, 1–36. [Google Scholar]
- Grosse, S.D.; A Boyle, C.; Botkin, J.R.; Comeau, A.M.; Kharrazi, M.; Rosenfeld, M.; Wilfond, B.S. Newborn screening for cystic fibrosis: Evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm. Rep. 2004, 53, 1–36. [Google Scholar] [PubMed]
- Henry, R.; Boulton, T.; Roddick, L. False negative results on newborn screening for cystic fibrosis. J. Paediatr. Child Health 1990, 26, 150–151. [Google Scholar] [CrossRef] [PubMed]
- MacLean, J.; Solomon, M.; Corey, M.; Selvadurai, H. Cystic fibrosis newborn screening does not delay the identification of cystic fibrosis in children with negative results. J. Cyst. Fibros. 2011, 10, 333–337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sosnay, P.R.; White, T.B.; Farrell, P.M.; Ren, C.L.; Derichs, N.; Howenstine, M.S.; Nick, J.A.; De Boeck, K. Diagnosis of cystic fibrosis in non screened populations. J. Pediatr. 2017, 181, S52–S57. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.; Farrell, P. Estimating the annual number of false negative cystic fibrosis newborn screening tests. Pediatr. Pulmonol. 2011, 47, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Rock, M.J.; Levy, H.; Zaleski, C.; Farrell, P.M. Factors accounting for a missed diagnosis of cystic fibrosis after newborn screening. Pediatr. Pulmonol. 2011, 46, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.; Sommerburg, O. Toward quality improvement in cystic fibrosis newborn screening: Progress and continuing challenges. J. Cyst. Fibros. 2016, 15, 267–269. [Google Scholar] [CrossRef]
- Heidendael, J.F.; Tabbers, M.M.; De Vreede, I. False negative newborn screen and neonatal cholestasis in a premature child with cystic fibrosis. Eur. J. Nucl. Med. Mol. Imaging 2013, 173, 1581–1583. [Google Scholar] [CrossRef]
- Collaco, J.M.; Panny, S.R.; Hamosh, A.; Mogayzel, P.J., Jr. False Negative Cystic Fibrosis Newborn Screen. Clin. Pediatr. 2010, 49, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.T.; Skrypek, M.M.; Powers, A.L.R.; Laguna, T.A. The Need for Vigilance: The Case of a False-Negative Newborn Screen for Cystic Fibrosis. Pediatrics 2011, 128, e446–e449. [Google Scholar] [CrossRef] [PubMed]
- Padoan, R.; Genoni, S.; Moretti, E.; Seia, M.; Giunta, A.; Corbetta, C. Genetic and clinical features of false-negative infants in a neonatal screening programme for cystic fibrosis. Acta Paediatr. 2002, 91, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Calvin, J.; Hogg, S.L.; McShane, D.; McAuley, S.A.; Iles, R.; Ross-Russell, R.; MacLean, F.M.; E Heeley, M.; Heeley, A.F. Thirty-years of screening for cystic fibrosis in East Anglia. Arch. Dis. Child. 2012, 97, 1043–1047. [Google Scholar] [CrossRef]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J. Diagnostic Sweat Testing: The Cystic Fibrosis Foundation Guidelines. J. Pediatr. 2007, 151, 85–89. [Google Scholar] [CrossRef]
- Festini, F.; Taccetti, G.; Galici, V.; Campana, S.; Mergni, G.; Repetto, T. Long-term health outcomes of neonatal screening for cystic fibrosis. Arch. Dis. Child. 2008, 93, 357–358. [Google Scholar] [CrossRef]
- Ryckman, K.K.; Berberich, S.L.; Shchelochkov, O.A.; Cook, D.E.; Murray, J.C. Clinical and environmental influences on metabolic biomarkers collected for newborn screening. Clin. Biochem. 2012, 46, 133–138. [Google Scholar] [CrossRef]
- Kloosterboer, M.; Hoffman, G.; Gershan, W.M.; Laxova, A.; Li, Z.; Farrell, P.; Rock, M. Clarification of Laboratory and Clinical Variables That Influence Cystic Fibrosis Newborn Screening with Initial Analysis of Immunoreactive Trypsinogen. Pediatrics 2009, 123, e338–e346. [Google Scholar] [CrossRef]
- Terlizzi, V.; Mergni, G.; Buzzetti, R.; Centrone, C.; Zavataro, L.; Braggion, C. Cystic fibrosis screen positive inconclusive diagnosis (CFSPIS): Experience in Tuscany, Italy. J. Cyst. Fibros. 2019, 18, 484–490. [Google Scholar] [CrossRef]
- Munck, A.; Mayell, S.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713. [Google Scholar] [CrossRef]
- Morinville, V.D.; Husain, S.Z.; Bai, H.; Barth, B.; Alhosh, R.; Durie, P.R.; Freedman, S.D.; Himes, R.W.; Lowe, M.E.; Pohl, J.; et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Rusakow, L.S.; Abman, S.H.; Sokol, R.J.; Seltzer, W.; Hammond, K.B.; Accurso, F.J. Immunoreactive trypsinogen levels in infants with cystic fibrosis complicated by meconium ileus. Screening 1993, 2, 13–17. [Google Scholar] [CrossRef]
- Southern, K.W.; Mérelle, M.M.; Dankert-Roelse, J.E.; Nagelkerke, A.D. Newborn screening for cystic fibrosis. Cochrane. Database Syst. Rev. 2009, 21, CD001402. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Sontag, M.K.; Wagener, J.S. Complications associated with symptomatic diagnosis in infants with cystic fibrosis. J. Pediatr. 2005, 147, S37–S41. [Google Scholar] [CrossRef]
- Ren, C.; Borowitz, D.; Gonska, T.; Howenstine, M.S.; Levy, H.; Massie, J.; Milla, C.; Munck, A.; Southern, K.W. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J. Pediatr. 2017, 181, S45–S51. [Google Scholar] [CrossRef] [PubMed]
- Tluczek, A.; Mischler, E.H.; Farrell, P.M.; Fost, N.; Peterson, N.M.; Carey, P.; Bruns, W.T.; McCarthy, C. Parents’ Knowledge of Neonatal Screening and Response to False-Positive Cystic Fibrosis Testing. J. Dev. Behav. Pediatr. 1992, 13, 181–186. [Google Scholar] [CrossRef]
- Gibson, L.; Cooke, R. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959, 23, 545–549. [Google Scholar] [PubMed]
- Gottlieb, R.P. Metabolic alkalosis in cystic fibrosis. J. Pediatr. 1971, 79, 930–936. [Google Scholar] [CrossRef]
- Terlizzi, V.; Di Lullo, A.M.; Comegna, M.; Centrone, C.; Pelo, E.; Castaldo, G.; Raia, V.; Braggion, C. S737F is a new CFTR mutation typical of patients originally from the Tuscany region in Italy. Ital. J. Pediatr. 2018, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Mergni, G.; Centrone, C.; Festini, F.; Taccetti, G. Trend of sweat chloride values in a cohort of patients carrying CFTR mutations of varying clinical consequence: Is there a risk of increasing sweat chloride over time? Pediatr. Pulmonol. 2020, 55, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Rendine, S.; Calafell, F.; Cappello, N.; Gagliardini, R.; Caramia, G.; Rigillo, N.; Silvetti, M.; Zanda, M.; Miano, A.; Battistini, F.; et al. Genetic history of cystic fibrosis mutations in Italy. Reg. Distrib. Ann. Hum. Genet. 1997, 61, 411–424. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| IRT1/IRT2 + LACT (1992–2010) | IRT1/IRT2 + LACT + IRT1/DNA (2011–2018) | Total | |
|---|---|---|---|
| Newborns screened for CF | 514,841 | 231,845 | 746,686 |
| Newborns with negative screening | 513,619 | 231,227 | 744,846 |
| Non-CF newborns with negative screening (true negative) | 513,603 | 231,225 | 744,828 |
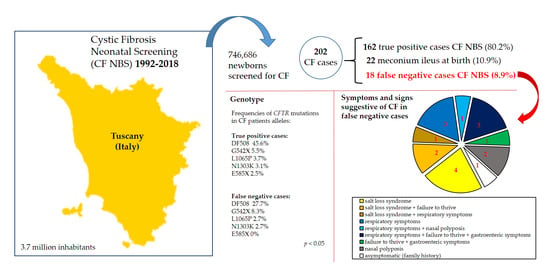
| CF newborns with negative screening (false negative) | 16 (11.2%) | 2 (3.4%) | 18 (8.9%) |
| Number of sweat tests (newborns with positive screening) | 1222 | 618 | 1840 |
| CF newborns with positive screening (true positive) | 112 (78.3%) | 50 (84.7%) | 162 (80.2%) |
| Non-CF newborns with positive screening (false positive) | 1110 | 568 | 1678 |
| CF newborns diagnosed with meconium ileus | 15 (10.5%) | 7 (11.9%) | 22 (10.9%) |
| Sensitivity (%) | 87.50 | 96.15 | 90.00 |
| Specificity (%) | 99.78 | 99.75 | 99.77 |
| Positive predictive value (%) | 9.16 | 8.09 | 8.80 |
| Negative predictive value (%) | 99.997 | 99.999 | 99.997 |
| FN Patients | Year of Birth | IRT1 µg/L (Cutoff) | CFTR Mutation 1 | CFTR Mutation 2 | Symptoms and Signs Suggestive of CF | Age at Diagnosis | Pancreas Status | Sweat Test (Chloride mEq) |
|---|---|---|---|---|---|---|---|---|
| A | 1993 | <55 (55) | G542X | D110H | asymptomatic (family history) | 22 years | PS | 89-80 |
| B | 1995 | <58 (58) | DF508 | E831X | nasal polyposis | 20 years | PS | 104-96 |
| C | 1996 | <58 (58) | DF508 | unknown | respiratory | 20 years | PS | 58-42-72-83 |
| D | 1997 | <60 (60) | G542X | unknown | respiratory + nasal polyposis | 17 years | PS | 62-67 |
| E | 1999 | <58 (58) | DF508 | 5T/12TG | respiratory | 15 years | PS | 78-100 |
| F | 1999 | <58 (58) | DF508 | unknown | failure to thrive + gastroenteric | 4 months | PD | 110-115 |
| G | 2001 | 39 (54) | DF508 | G542X | nasal polyposis | 5 years | PD | 80-94 |
| H | 2001 | 37 (54) | S737F | Del exon 22–24 | salt-loss syndrome + respiratory | 1 year | PS | 82-89 |
| I | 2003 | 48 (54) | DF508 | DF508 | respiratory + failure to thrive + gastroenteric | 4 months | PD | 110-153 |
| L | 2003 | 45 (54) | W1282X | DF311 | salt-loss syndrome | 5 months | PS | 60-63-85-93 |
| M | 2005 | 47 (62) | DF311 | Del exon 2 | salt-loss syndrome + failure to thrive | 4 months | PS | 75-71 |
| N | 2006 | 52 (57) | L206W | 3659delC | salt-loss syndrome | 10 months | PS | 90-65-156 |
| O | 2006 | 41 (57) | L1065P | 5T/13TG | respiratory | 12 years | PS | 52-61-40-69-101 |
| P | 2008 | 31 (60) | S737F | S737F | salt-loss syndrome | 7 months | PS | 45-46-93-64-98 |
| Q | 2010 | 51 (63) | DF508 | DF508 | respiratory + failure to thrive + gastroenteric | 4 months | PD | 102-96 |
| R | 2010 | 54 (63) | DF508 | Delexon 22–24 | respiratory + failure to thrive + gastroenteric | 10 months | PD | 125-125 |
| S | 2015 | 15 (50) | E831X | G126D | salt-loss syndrome | 11 months | PS | 67-74-70 |
| T | 2016 | 27 (49) | N1303K | L997F | salt-loss syndrome + failure to thrive | 11 months | PS | 60-58-84-67 |
| True-Positive Cases | False-Negative Cases | ||
|---|---|---|---|
| Mutations | Frequency (%) | Mutations | Frequency (%) |
| DF508 | 45.6 | DF508 | 27.7 |
| G542X | 5.5 | G542X | 8.3 |
| L1065P | 3.7 | S737F | 8.3 |
| N1303K | 3.1 | Delexon 22–24 | 5.5 |
| E585X | 2.5 | L1065P | 2.7 |
| 2789+5G->A | 2.5 | W1282X | 2.7 |
| Delexon 22–24 | 2.2 | N1303K | 2.7 |
| R347P | 2.2 | Other rare mutations | 33.3 |
| W1282X | 2.2 | Unknown | 8.3 |
| T338I | 2.2 | ||
| R553X | 1.8 | ||
| S737F | 1.3 | ||
| 2183AA->G | 1.3 | ||
| Other rare mutations | 22.8 | ||
| Unknown | 1.1 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taccetti, G.; Botti, M.; Terlizzi, V.; Cavicchi, M.C.; Neri, A.S.; Galici, V.; Mergni, G.; Centrone, C.; Peroni, D.G.; Festini, F. Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy. Diagnostics 2020, 10, 446. https://doi.org/10.3390/diagnostics10070446
Taccetti G, Botti M, Terlizzi V, Cavicchi MC, Neri AS, Galici V, Mergni G, Centrone C, Peroni DG, Festini F. Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy. Diagnostics. 2020; 10(7):446. https://doi.org/10.3390/diagnostics10070446
Chicago/Turabian StyleTaccetti, Giovanni, Matteo Botti, Vito Terlizzi, Maria Chiara Cavicchi, Anna Silvia Neri, Valeria Galici, Gianfranco Mergni, Claudia Centrone, Diego G. Peroni, and Filippo Festini. 2020. "Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy" Diagnostics 10, no. 7: 446. https://doi.org/10.3390/diagnostics10070446
APA StyleTaccetti, G., Botti, M., Terlizzi, V., Cavicchi, M. C., Neri, A. S., Galici, V., Mergni, G., Centrone, C., Peroni, D. G., & Festini, F. (2020). Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy. Diagnostics, 10(7), 446. https://doi.org/10.3390/diagnostics10070446